商品详情
返回产品目录
商品包装及说明书因厂家更换频繁,如有不符以实物为主
维奈克拉片
国际零售参考价:¥**/盒
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。
1 适应症及用途
1.1 慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
VENCLEXTA 适用于治疗患有慢性淋巴细胞白血病(CLL)或小淋巴细胞淋巴瘤(SLL)的成年患者。
1.2 急性粒细胞白血病
VENCLEXTA 与阿扎胞苷、地西他滨或低剂量阿糖胞苷联合用于治疗 75 岁或以上成人新诊断的急性髓系白血病 (AML),或患有无法使用强化诱导化疗的合并症的患者。
2 剂量和给药方法
2.1 重要安全信息
评估肿瘤溶解综合征(TLS)风险水平的患者特异性因素和在首次剂量VENCLEXTA之前向患者提供预防性水化和抗高尿酸血症以降低TLS风险[见剂量和给药方法(2.4)和警告和注意事项( 5.1)]。
2.2 慢性淋巴细胞白血病/小淋巴细胞淋巴瘤的推荐剂量
VENCLEXTA 剂量从 5 周的加量开始。5 周的剂量递增方案旨在逐渐减少肿瘤负荷(减瘤)并降低 TLS 风险。
VENCLEXTA 5 周剂量递增时间表
根据 5 周剂量增加方案给予 VENCLEXTA 至推荐剂量 400 mg 口服每日一次,如表 1所示。
表 1. CLL/SLL 患者 5 周加速阶段的给药方案
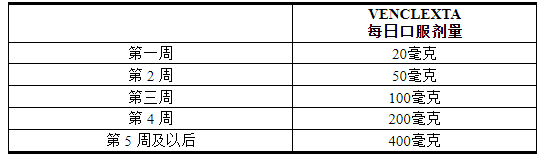
CLL/SLL 起始包根据启动计划提供前 4 周的 VENCLEXTA [请参阅如何提供/储存和处理 ( 16 )]。
与奥比妥珠单抗联用
在第 1 周期第 1 天开始给予 obinutuzumab 100 mg,随后在第 1 周期第 2 天给予 900 mg。在第 1 周期第 8 和 15 天以及每个后续 28 天周期的第 1 天给予 1000 mg,总共 6 个周期。有关其他剂量信息,请参阅 obinutuzumab 处方信息。
在第 1 周期第 22 天,根据 5 周递增给药方案开始 VENCLEXTA(见表1)。在第 2 周期第 28 天完成加速阶段后,从第 3 周期第 1 天直至第 12 周期最后一天,继续以 400 mg 剂量口服 VENCLEXTA,每天一次。
与利妥昔单抗联合使用
在患者完成 VENCLEXTA 的 5 周加速给药方案(见表1)并接受每日一次 400 mg 口服推荐剂量的 VENCLEXTA 治疗 7 天后,开始利妥昔单抗给药。在每个28天周期的第1天施用利妥昔单抗,共6个周期,第1周期的剂量为375 mg/m 2静脉注射,第2-6周期的静脉注射剂量为500 mg/m 2。从利妥昔单抗第 1 周期第 1 天开始,继续每日口服 VENCLEXTA 400 mg,持续 24 个月。
有关其他剂量信息,请参阅利妥昔单抗处方信息。
单一疗法
完成 5 周的剂量递增方案后,VENCLEXTA 的推荐剂量为 400 mg 每日一次(见表1)。继续使用 VENCLEXTA 直至疾病进展或出现不可接受的毒性。
2.3 急性髓系白血病的推荐剂量
VENCLEXTA 的推荐剂量和增量取决于联合用药。遵循给药方案,包括 3 天或 4 天剂量递增,如表 2 所示。在第 1 周期第 1 天开始 VENCLEXTA 给药,并与以下药物组合:
- 阿扎胞苷 75 mg/m 2静脉注射或皮下注射,每天一次,每个 28 天周期的第 1-7 天;或者
- 地西他滨 20 mg/m 2静脉注射,每天一次,每个 28 天周期的第 1-5 天;或者
- 阿糖胞苷 20 mg/m 2皮下注射,每天一次,每个 28 天周期的第 1-10 天。
表 2. AML 患者 3 天或 4 天加速阶段的给药方案
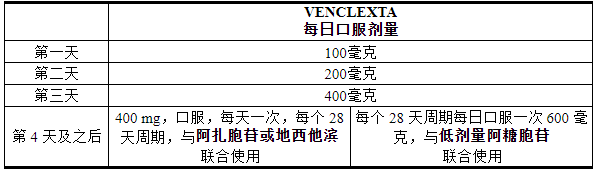
继续使用 VENCLEXTA 与阿扎胞苷或地西他滨或低剂量阿糖胞苷联用,直至疾病进展或出现不可接受的毒性。
有关其他剂量信息,请参阅阿扎胞苷、地西他滨或阿糖胞苷的临床研究 ( 14.2 )和处方信息。
2.4 肿瘤溶解综合征的风险评估和预防
用 VENCLEXTA 治疗的患者可能会出现肿瘤溶解综合征 (TLS)。有关管理的具体细节,请参阅下面的相应部分。评估患者特定因素的 TLS 风险水平,并在首次服用 VENCLEXTA 之前向患者提供预防性水合和抗高尿酸血症药物,以降低 TLS 风险。
慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
VENCLEXTA 可导致肿瘤快速缩小,因此在最初的 5 周加速阶段存在 TLS 风险。首次服用 VENCLEXTA 后 6 至 8 小时以及每次剂量增加时,可能会出现与 TLS 一致的血液化学变化,需要及时处理。剂量中断后恢复 VENCLEXTA 时也可能发生 TLS。中断后 VENCLEXTA 的剂量修改参见表4和表 5 。
TLS 的风险是基于多种因素的连续体,特别是肾功能下降(肌酐清除率 [CLcr] <80 mL/min)和肿瘤负荷;脾肿大也可能增加 TLS 的风险。
在开始使用 VENCLEXTA 治疗之前,进行肿瘤负荷评估,包括放射学评估(例如 CT 扫描),评估所有患者的血液化学(钾、尿酸、磷、钙和肌酐)并纠正预先存在的异常。随着肿瘤负荷降低,风险可能降低[参见警告和注意事项( 5.1 )和特殊人群中的使用( 8.6 )]。
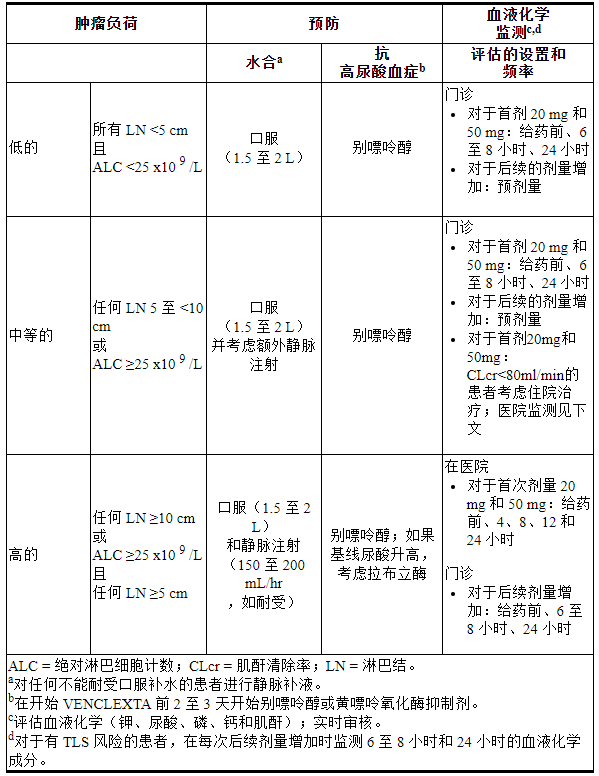
下表3描述了根据临床试验数据确定的肿瘤负荷在VENCLEXTA治疗期间推荐的TLS预防和监测。在最终确定预防和监测计划之前,请考虑所有患者的合并症。在加速阶段剂量中断持续超过 1 周或完成加速后超过 2 周后重新开始 VENCLEXTA 时,重新评估 TLS 风险。根据需要进行预防和监测。
表 3. 根据 CLL/SLL 患者的肿瘤负荷推荐的 TLS 预防
急性髓系白血病
- 在开始 VENCLEXTA 之前,所有患者的白细胞计数应低于 25 × 10 9 /L。治疗前可能需要进行细胞减灭术。
- 在第一次 VENCLEXTA 剂量之前,为所有患者提供预防措施,包括充足的水合和抗高尿酸血症药物,并在加速阶段继续进行。
- 在开始使用 VENCLEXTA 治疗之前,评估血液化学(钾、尿酸、磷、钙和肌酐)并纠正先前存在的异常。
- 在给药前、每次新剂量后 6 至 8 小时以及达到最终剂量后 24 小时监测 TLS 的血液化学成分。
- 对于有 TLS 危险因素(例如循环母细胞、骨髓白血病负担高、治疗前乳酸脱氢酶 [LDH] 水平升高或肾功能下降)的患者,考虑其他措施,包括加强实验室监测和减少 VENCLEXTA 起始剂量。
2.5 不良反应的剂量调整
慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
表4中提供了针对不良反应的VENCLEXTA推荐剂量修改,表5中提供了针对不良反应的VENCLEXTA推荐剂量减少。
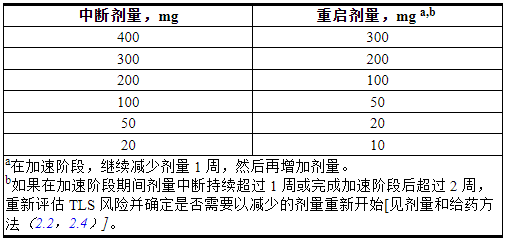
对于在加速阶段期间剂量中断持续超过 1 周或完成加速阶段后超过 2 周的患者,重新评估 TLS 风险,以确定是否需要重新开始减少剂量(例如,全部或剂量增加计划的某些水平)[见剂量和给药方法(2.2,2.4 ) ]。
表 4. CLL/SLL 中不良反应的推荐 VENCLEXTA 剂量修改
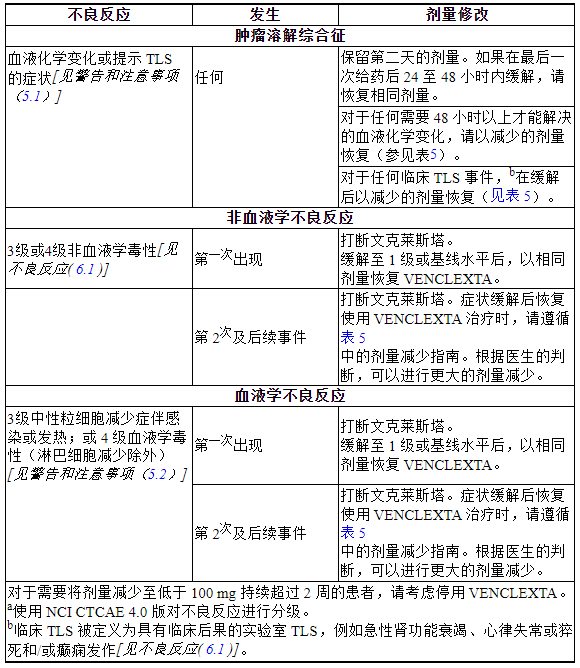
表 5. CLL/SLL 中 VENCLEXTA 不良反应的推荐剂量减少
急性髓系白血病
通过解决血细胞减少症经常监测血细胞计数。血细胞减少症的剂量调整和中断取决于缓解状态。表6中提供了针对不良反应的VENCLEXTA剂量修改。
表 6. 针对 AML 不良反应推荐的 VENCLEXTA 剂量修改

2.6 药物相互作用的剂量调整
强或中度 CYP3A 抑制剂或 P-gp 抑制剂
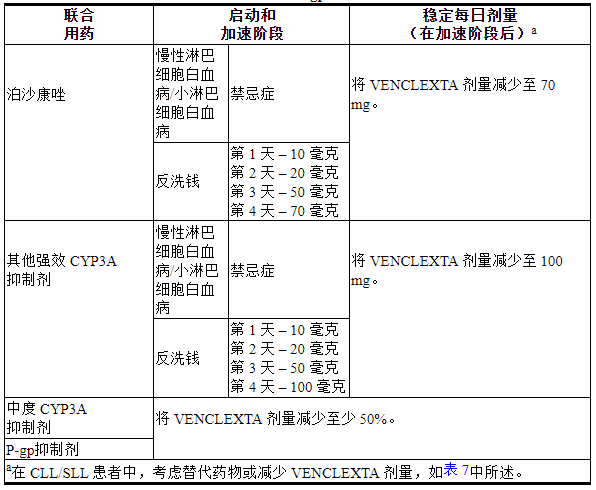
表7描述了基于与强或中度CYP3A抑制剂或P-gp抑制剂同时使用的VENCLEXTA禁忌症或剂量修改[见药物相互作用( 7.1 )]在开始、期间或加速期之后。
终止抑制剂后2至3天恢复在同时使用强或中度CYP3A抑制剂或P-gp抑制剂之前使用的VENCLEXTA剂量[见药物相互作用( 7.1 ) ]。
表 7. VENCLEXTA 与 CYP3A 和 P-gp 抑制剂潜在相互作用的处理
2.7 严重肝受损患者的剂量调整
对于严重肝受损患者(Child-Pugh C),每日一次 VENCLEXTA 剂量减少 50%;更密切地监测这些患者的不良反应[见特殊人群中的使用( 8.7 )]。
2.8 管理
指导患者以下事项:
- 与膳食和水一起服用 VENCLEXTA。
- 每天大约在同一时间服用 VENCLEXTA。
- 整个吞服 VENCLEXTA 片剂。吞咽前请勿咀嚼、压碎或打破药片。
VENCLEXTA 的推荐剂量可以使用任何批准的片剂规格进行递送(例如,患者可以根据需要服用 2 x 50 mg 片剂或 10 x 10 mg 片剂,而不是 1 x 100 mg 片剂)。
如果患者在通常服用时间的8小时内漏服一剂VENCLEXTA,指导患者尽快服用漏服的剂量并恢复正常的每日给药时间表。如果患者错过服药时间超过 8 小时,请指导患者不要服用错过的剂量,并在第二天恢复通常的给药时间表。
如果患者在服药后呕吐,指导患者当天不要服用额外的剂量,并在通常的时间服用下一个处方剂量。
3 剂型和规格
表 8. VENCLEXTA 片剂强度和描述

4 禁忌症
在有 CLL/SLL 患者中,在开始和加速阶段同时使用 VENCLEXTA 与强CYP3A 抑制剂是禁忌的,因为肿瘤溶解综合征风险可能增加[见剂量和给药方法( 2.6 )和药物相互作用( 7.1 ) ]。
5 警告和注意事项
5.1 肿瘤溶解综合征
用VENCLEXTA治疗患者曾发生肿瘤溶解综合征(TLS),包括致命事件和需要透析的肾衰竭[见不良反应( 6.1 )]。
VENCLEXTA 可导致肿瘤快速缩小,因此在所有患者的起始阶段和加速阶段以及 CLL/SLL 患者的剂量中断后重新开始期间存在 TLS 风险。首次服用 VENCLEXTA 后 6 至 8 小时以及每次剂量增加时,可能会出现与 TLS 一致的血液化学变化,需要及时处理。单次 20 mg 剂量的 VENCLEXTA 后已有 TLS 病例报告,包括致命病例。
在接受当前(5周)剂量增加以及 TLS 预防和监测措施的 CLL/SLL 患者中,VENCLEXTA CLL/SLL 单药治疗试验中 TLS 发生率为 2%。TLS 率与 VENCLEXTA 联合 obinutuzumab 或 rituximab 保持一致。在有 CLL/SLL 患者中进行 2 至 3 周剂量增加和较高起始剂量时,TLS 率为 13%,包括死亡和肾衰竭[见不良反应( 6.1 )]。
在遵循当前 3 天剂量递增方案以及 TLS 预防和监测措施的 AML 患者中,接受 VENCLEXTA 联合阿扎胞苷 (VIALE-A) 治疗的患者 TLS 发生率为 1.1%。在遵循 4 天剂量递增方案以及 TLS 预防和监测措施的 AML 患者中,接受 VENCLEXTA 联合低剂量阿糖胞苷 (VIALE) 治疗的患者中,TLS 发生率为 5.6%,其中包括死亡和肾功能衰竭-C) [参见不良反应 ( 6.1 )]。
TLS 的风险是基于多种因素的连续体,特别是肾功能下降、肿瘤负荷和恶性肿瘤类型。脾肿大也可能增加 CLL/SLL 患者发生 TLS 的风险。
评估所有患者的风险并提供适当的 TLS 预防措施,包括水合和抗高尿酸血症。监测血液化学并及时处理异常情况。随着总体风险的增加,采取更强化的措施(静脉补液、频繁监测、住院治疗)。如果需要,中断给药;当重新启动VENCLEXTA时,遵循剂量修改指南[见剂量和给药方法( 2.1,2.2,2.3,2.4 )和在特殊人群中使用( 8.6 ) ]。
VENCLEXTA 与 P-gp 抑制剂或强或中度 CYP3A 抑制剂同时使用会增加Venetoclax暴露,这可能会增加 VENCLEXTA 起始和加速阶段发生 TLS 的风险。对于有CLL/SLL患者,在开始时和5周加速阶段期间VENCLEXTA与强CYP3A抑制剂的共同给药是禁忌的[见禁忌证( 4 )]。对于 AML 患者,在开始时和 3 天或 4 天的加速阶段期间与强效 CYP3A 抑制剂共同给药时,减少 VENCLEXTA 的剂量。对于有CLL/SLL或AML患者,当与中度CYP3A4抑制剂或P-gp抑制剂共同给药时减少VENCLEXTA的剂量[见剂量和给药方法( 2.6))和药物相互作用(7.1)]。
5.2 中性粒细胞减少症
在 CLL 患者中,在联合治疗和单药治疗研究中用 VENCLEXTA 治疗时,63% 至 64% 的患者出现 3 级或 4 级中性粒细胞减少症,31% 至 33% 的患者出现 4 级中性粒细胞减少症。4%至6%的患者发生发热性中性粒细胞减少症[见不良反应( 6.1 )]。
在 AML 患者中,接受 VENCLEXTA 联合阿扎胞苷、地西他滨或低剂量阿糖胞苷治疗的患者中 95% 至 100% 的基线中性粒细胞计数恶化。中性粒细胞减少症可能会在后续周期中复发。
在整个治疗期间监测全血细胞计数。对于VENCLEXTA对严重中性粒细胞减少症的中断和剂量恢复,对于CLL 见表4和对于AML表6 [见剂量和给药方法( 2.5 )]。考虑支持性措施,包括抗菌药物和生长因子(例如 G-CSF)。
5.3 感染
用VENCLEXTA治疗患者曾发生致命和严重感染,如肺炎和败血症[见不良反应( 6.1 )]。
监测患者的感染体征和症状并及时治疗。对于 3 级和 4 级感染,暂停使用 VENCLEXTA 直至解决。对于剂量恢复,对于CLL参见表4和对于AML参见表6 [参见剂量和给药方法( 2.5 )]。
5.4 免疫接种
在使用 VENCLEXTA 治疗之前、期间或之后,请勿接种减毒活疫苗,直至 B 细胞恢复。尚未研究 VENCLEXTA 治疗期间或之后使用减毒活疫苗免疫的安全性和有效性。告知患者疫苗接种的效果可能较差。
5.5 胚胎-胎儿毒性
根据动物研究结果及其作用机制,VENCLEXTA 在给孕妇服用时可能会造成胚胎-胎儿伤害。在小鼠中进行的一项胚胎-胎儿研究中,给怀孕动物施用Venetoclax的暴露量相当于患者每天 400 mg 剂量所观察到的暴露量,导致着床后损失和胎儿体重减轻。
告知孕妇对胎儿的潜在风险。忠告有生殖潜力的女性用VENCLEXTA治疗期间和末次剂量后共30天使用有效避孕[见特殊人群中使用( 8.1,8.3 )] 。
5.6 当 VENCLEXTA 添加至硼替佐米和地塞米松时,多发性骨髓瘤患者的死亡率增加
在一项针对复发性或难治性多发性骨髓瘤患者的随机试验(BELLINI;NCT02755597)中,在硼替佐米加地塞米松(VENCLEXTA 不适用的用途)中添加 VENCLEXTA 导致死亡率增加。在对照临床试验之外,不推荐使用 VENCLEXTA 联合硼替佐米加地塞米松治疗多发性骨髓瘤患者。
6 不良反应
标签中其他地方描述了以下具有临床意义的不良反应:
- 肿瘤溶解综合征[参见警告和注意事项 ( 5.1 )]
- 中性粒细胞减少症[参见警告和注意事项(5.2)]
- 感染[参见警告和注意事项(5.3)]
6.1 临床试验经验
由于临床试验是在广泛变化的条件下进行的,因此在一种药物的临床试验中观察到的不良事件发生率不能直接与另一种药物的临床试验的发生率进行比较,并且可能无法反映在实践中观察到的发生率。
在CLL/SLL中,安全性人群反映了M13-982、M14-032和M12-175患者中VENCLEXTA作为单药治疗以及CLL14和MURANO患者中与obinutuzumab或利妥昔单抗联用的暴露。在这个 CLL/SLL 安全人群中,VENCLEXTA 最常见的不良反应(≥20%)是中性粒细胞减少、血小板减少、贫血、腹泻、恶心、上呼吸道感染、咳嗽、肌肉骨骼疼痛、疲劳和水肿。
在 AML 中,安全人群反映了 M14-358、VIALE-A 和 VIALE-C 患者中 VENCLEXTA 与地西他滨、阿扎胞苷或低剂量阿糖胞苷联用的暴露。在这个安全人群中,最常见的不良反应(任何试验中≥30%)是恶心、腹泻、血小板减少、便秘、中性粒细胞减少、发热性中性粒细胞减少、疲劳、呕吐、水肿、发热、肺炎、呼吸困难、出血、贫血、皮疹、腹痛、败血症、肌肉骨骼疼痛、头晕、咳嗽、口咽痛和低血压。
慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
VENCLEXTA 与 Obinutuzumab 联用
在 CLL14 中评估了 VENCLEXTA 联合 obinutuzumab (VEN+G) (N=212) 与 obinutuzumab 联合苯丁酸氮芥 (GClb) (N=214) 的安全性,CLL14 是一项针对患有以下疾病的患者的随机、开放标签、主动对照试验:既往未经治疗的 CLL [参见临床研究 ( 14.1 )]。随机分配至 VEN+G 组的患者接受 VENCLEXTA 和 obinutuzumab 联合治疗六个周期,然后使用 VENCLEXTA 作为单一疗法再进行六个周期。患者在第 1 个周期的第 22 天开始 VENCLEXTA 5 周加速的第一剂,完成后继续每日一次口服 VENCLEXTA 400 mg,总共 12 个周期。该试验要求总累积疾病评定量表 (CIRS) >6 或 CLcr <70 mL/min,肝转氨酶和总胆红素≤正常上限的 2 倍,并排除 CIRS 任何个体器官/系统损伤评分为 4 分的患者,除外眼、耳、鼻、喉器官系统。VEN+G 组中暴露于 VENCLEXTA 的中位持续时间为 10.5 个月(范围:0 至 13.5 个月),obinutuzumab 的中位周期数为 6。
VEN+G 组中 49% 的患者报告出现严重不良反应,最常见的是发热性中性粒细胞减少症和肺炎(各 5%)。据报道,2% (4/212) 的患者在疾病没有进展的情况下以及在上次研究治疗后 28 天内发生致命不良反应,最常见的是感染。
在 VEN+G 组中,不良反应导致 16% 的患者停止治疗,21% 的患者减少剂量,74% 的患者中断剂量。中性粒细胞减少症导致 2% 的患者停用 VENCLEXTA,13% 的患者剂量减少,41% 的患者剂量中断。
表 9列出了 CLL14 中发现的不良反应。
表 9. CLL14 中接受 VEN+G 治疗的患者的不良反应 (≥10%)
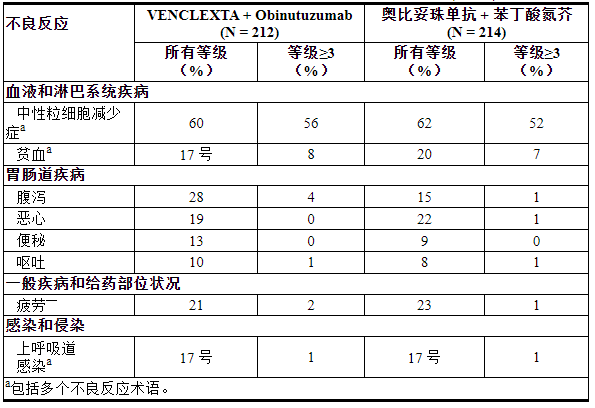
在接受 VEN+G 治疗的患者中报告的其他临床重要不良反应(所有级别)低于 10%,如下所示:
血液和淋巴系统疾病:发热性中性粒细胞减少症(6%)
感染和侵染(均包括多种不良反应术语):肺炎(9%)、尿路感染(6%)、败血症(4%)
代谢和营养障碍:肿瘤溶解综合征(1%)
在VEN+G完成后接受VENCLEXTA单药治疗期间,≥10%的患者发生的不良反应是中性粒细胞减少症(26%)。≥2%的患者发生的≥3级不良反应为中性粒细胞减少(23%)和贫血(2%)。
表 10列出了 CLL14 的实验室异常情况。
表 10. CLL14 中接受 VEN+G 治疗的患者出现新的或恶化的临床重要实验室异常 (≥10%)
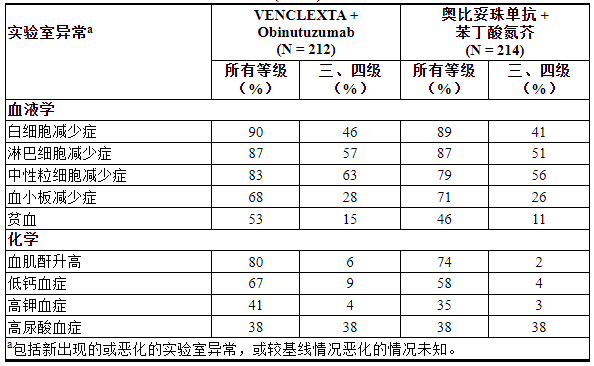
≥2%接受VEN+G治疗的患者出现4级实验室异常,包括中性粒细胞减少症(32%)、白细胞减少症和淋巴细胞减少症(10%)、血小板减少症(8%)、低钙血症(8%)、高尿酸血症(7%)、血肌酐升高(3%)、高钙血症(3%)和低钾血症(2%)。
VENCLEXTA 与利妥昔单抗联用
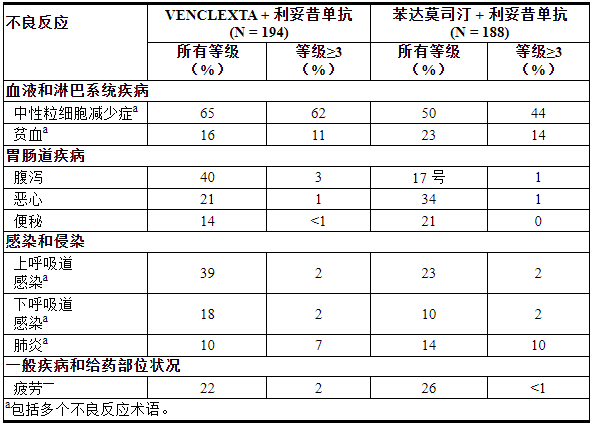
在MURANO中评价VENCLEXTA联用利妥昔单抗(VEN+R)(N=194)相对于苯达莫司汀联用利妥昔单抗(B+R)(N=188)的安全性[见临床研究( 14.1 ) ]。随机接受 VEN+R 的患者完成了预定的加速期(5 周),接受 VENCLEXTA 400 mg 每日一次,联合利妥昔单抗治疗 6 个周期,随后接受 VENCLEXTA 单药治疗,加速期后总共 24 个月。在分析时,VEN+R 组中暴露于 VENCLEXTA 的中位持续时间为 22 个月,利妥昔单抗的中位周期数为 6 个。
VEN+R 组中有 46% 的患者报告出现严重不良反应,其中最常见(≥5%)的是肺炎(9%)。2% (4/194) 的患者报告在没有疾病进展的情况下以及最后一次 VENCLEXTA 治疗后 30 天内和/或最后一次利妥昔单抗治疗 90 天内发生致命不良反应。
在 VEN+R 组中,不良反应导致 16% 的患者停止治疗,15% 的患者减少剂量,71% 的患者中断剂量。中性粒细胞减少症和血小板减少症分别导致 3% 的患者停用 VENCLEXTA。46% 的患者因中性粒细胞减少症导致 VENCLEXTA 剂量中断。
表 11列出了 MURANO 中发现的不良反应。
表 11. MURANO 接受 VEN+R 治疗的患者的不良反应(≥10%)
在接受 VEN+R 治疗的患者中报告的其他临床重要不良反应(所有级别)低于 10%,如下所示:
血液和淋巴系统疾病:发热性中性粒细胞减少症(4%)
胃肠道疾病:呕吐(8%)
感染和侵染:败血症(<1%)
代谢和营养障碍:肿瘤溶解综合征(3%)
VEN+R联合治疗完成后,接受VENCLEXTA单药治疗期间,≥10%患者发生的不良反应为上呼吸道感染(21%)、腹泻(19%)、中性粒细胞减少(16%)和下呼吸道感染感染(11%)。≥2%的患者发生的3级或4级不良反应为中性粒细胞减少症(12%)和贫血(3%)。
表 12列出了 MURANO 中发现的实验室异常情况。
表 12. MURANO 接受 VEN+R 治疗的患者出现新的或恶化的临床重要实验室异常 (≥10%)
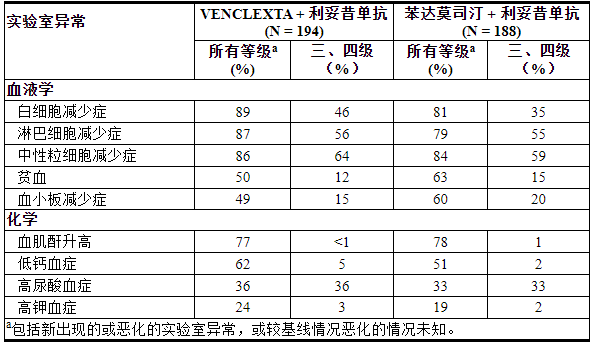
≥2%接受VEN+R治疗的患者出现4级实验室异常,包括中性粒细胞减少症(31%)、淋巴细胞减少症(16%)、白细胞减少症(6%)、血小板减少症(6%)、高尿酸血症(4%)、低钙血症( 2%)、低血糖(2%)和高镁血症(2%)。
VENCLEXTA 作为单一疗法
VENCLEXTA 的安全性通过三项单臂试验(M13-982、M14-032 和 M12-175)的汇总数据进行评估。完成加速阶段后,患者每天口服一次 VENCLEXTA 400 mg(N=352)。数据分析时,VENCLEXTA 治疗的中位持续时间为 14.5 个月(范围:0 至 50 个月)。52% 的患者接受 VENCLEXTA 治疗超过 60 周。
在汇总数据集中,中位年龄为 66 岁(范围:28 至 85 岁),93% 为白人,68% 为男性。先前治疗的中位数为 3 次(范围:0 至 15)。
52% 的患者报告出现严重不良反应,其中最常见(≥5%)的是肺炎(9%)、发热性中性粒细胞减少症(5%)和败血症(5%)。在 VENCLEXTA 单药治疗研究中,2% 的患者报告了在疾病无进展的情况下以及在Venetoclax治疗 30 天内发生的致命不良反应,最常见的是(2 名患者)来自脓毒性休克。
不良反应导致 9% 的患者停止治疗,13% 的患者减少剂量,36% 的患者中断剂量。导致停药的最常见不良反应是血小板减少症和自身免疫性溶血性贫血。导致剂量减少或中断的最常见不良反应(≥5%)是中性粒细胞减少症(8%)。
表 13列出了这些试验中发现的不良反应。
表 13. 接受 VENCLEXTA 单药治疗的既往治疗过的 CLL/SLL 患者中≥10%(所有级别)或≥5%(级别≥3)报告的不良反应
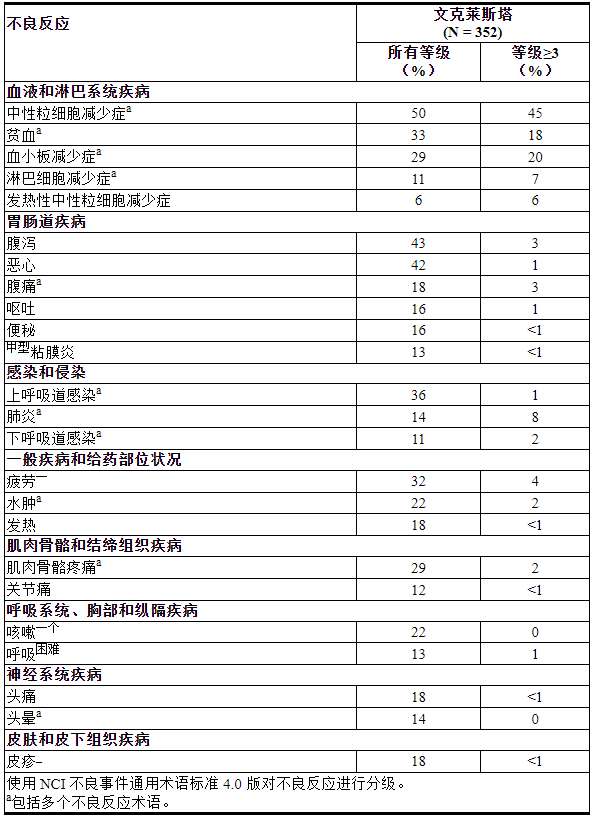
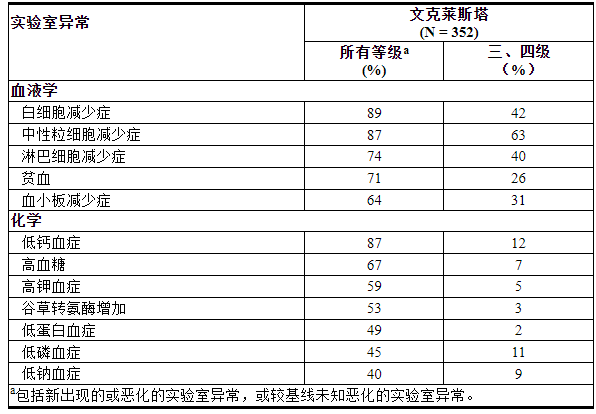
表 14 列出了整个治疗过程中报告的实验室异常情况,这些异常情况是新出现的或较基线恶化的。用 VENCLEXTA 单药治疗观察到的最常见 (>5%) 4 级实验室异常是血液学实验室异常,包括中性粒细胞减少 (33%)、白细胞减少 (11%)、血小板减少 (15%) 和淋巴细胞减少 (9%)。
表 14. 接受 VENCLEXTA 单药治疗的既往治疗过的 CLL/SLL 患者中≥40%(所有级别)或≥10%(3 级或 4 级)出现新的或恶化的实验室异常
CLL/SLL 的重要不良反应
肿瘤溶解综合征
肿瘤溶解综合征是开始使用 VENCLEXTA 时发现的一个重要风险。
慢性淋巴细胞白血病14
用VEN+G治疗患者中TLS的发生率为1%(3/212) [见警告和注意事项( 5.1 )]。TLS 的所有三个事件均得到解决,并且没有导致试验退出。由于 TLS 事件,Obinutuzumab 给药在两例病例中被延迟。
穆拉诺
在接受 VEN+R 治疗的患者中,TLS 的发生率为 3% (6/194)。在 77/389 名患者参加试验后,对方案进行了修订,纳入了第 2.2 和 2.4 节中描述的当前 TLS 预防和监测措施[见剂量和给药方法( 2.2 , 2.4 )]。所有 TLS 事件都发生在 VENCLEXTA 启动期间,并在两天内得到解决。所有 6 名患者均完成了剂量增加并达到 VENCLEXTA 每日推荐剂量 400 mg。在遵循当前 5 周加速计划和 TLS 预防和监测措施的患者中未观察到临床 TLS [见剂量和给药方法(2.2,2.4)]。表12列出了用VEN+R治疗的患者的与TLS相关的实验室异常率。
单药治疗研究(M13-982 和 M14-032)
在根据第2.1和2.2节中描述的建议治疗的168名CLL患者中, TLS的发生率为2% [见剂量和给药方法( 2.2,2.4 )]。所有事件均符合实验室 TLS 标准(实验室异常在 24 小时内满足以下至少 2 个条件:钾 >6 mmol/L、尿酸 >476 µmol/L、钙 <1.75 mmol/L 或磷 >1.5 mmol/L),或报告为 TLS 事件。这些事件发生在淋巴结≥5 cm 和/或绝对淋巴细胞计数 (ALC) ≥25 x 10 9的患者中/L。所有事件均在 5 天内得到解决。在这些患者中未观察到 TLS 导致急性肾功能衰竭、心律失常或猝死和/或癫痫发作等临床后果。所有患者的 CLcr ≥50 mL/min。与 TLS 相关的实验室异常包括高钾血症(17% 所有级别,1% ≥3 级)、高磷血症(14% 所有级别,2% ≥3 级)、低钙血症(16% 所有级别,2% ≥3 级)和高尿酸血症(10% 所有等级,<1% 等级≥3)。
在最初的 1 期剂量探索试验中,启动阶段较短(2-3 周),起始剂量较高,TLS 的发生率为 13%(10/77;5 例实验室 TLS,5 例临床 TLS),包括2 起致命事件和 3 起急性肾衰竭事件,其中 1 起需要透析。在此经验之后,修订了 TLS 风险评估、给药方案、TLS 预防和监测措施[见剂量和给药方法(2.2,2.4)]。
急性髓系白血病
VENCLEXTA 与阿扎胞苷联用
VIALE-A 是一项双盲、随机试验,评估了 VENCLEXTA 联合阿扎胞苷 (VEN+AZA) (N=283) 与安慰剂联合阿扎胞苷 (PBO+AZA) (N=144) 的安全性。新诊断的 AML 患者[参见临床研究(14.2)]。基线时,患者年龄≥75岁,或患有基于以下至少一项标准而无法使用强化诱导化疗的合并症:基线ECOG表现状态为2-3,严重心脏或肺部合并症,中度肝功能损害, CLcr <45 mL/min,或其他合并症。患者在完成加速阶段后随机接受 VENCLEXTA 400 mg 口服,每日一次,联合阿扎胞苷(75 mg/m 2每个 28 天周期的第 1-7 天静脉注射或皮下注射)或安慰剂与阿扎胞苷组合。在接受 VEN+AZA 的患者中,暴露于 VENCLEXTA 的中位持续时间为 7.6 个月(范围:<0.1 至 30.7 个月)。
接受 VEN+AZA 治疗的患者中有 83% 报告出现严重不良反应,其中最常见(≥5%)的是发热性中性粒细胞减少症(30%)、肺炎(22%)、败血症(不包括真菌;19%)和出血(6%)。接受 VEN+AZA 治疗的患者中有 23% 发生致命不良反应,其中最常见(≥2%)的是肺炎(4%)、败血症(不包括真菌;3%)和出血(2%)。
不良反应导致 24% 的患者永久停用 VENCLEXTA,2% 的患者减少剂量,72% 的患者中断剂量。导致≥2%患者停用VENCLEXTA的不良反应为脓毒症(不包括真菌;3%)和肺炎(2%)。导致剂量减少的最常见不良反应是肺炎(0.7%)。≥5%的患者需要中断剂量的不良反应包括发热性中性粒细胞减少症(20%)、中性粒细胞减少症(20%)、肺炎(14%)、败血症(不包括真菌;11%)和血小板减少症(10%)。在实现白血病骨髓清除的患者中,53% 的患者因绝对中性粒细胞计数 (ANC) <500/微升而接受剂量中断。
表 15列出了 VIALE-A 中发现的不良反应。
表 15. VIALE-A 中接受 VEN+AZA 治疗的 AML 患者的不良反应 (≥10%),与 VIALE-A 中的 PBO+AZA 相比,所有级别的组间差异≥5%,3 级或 4 级反应的组间差异≥2%
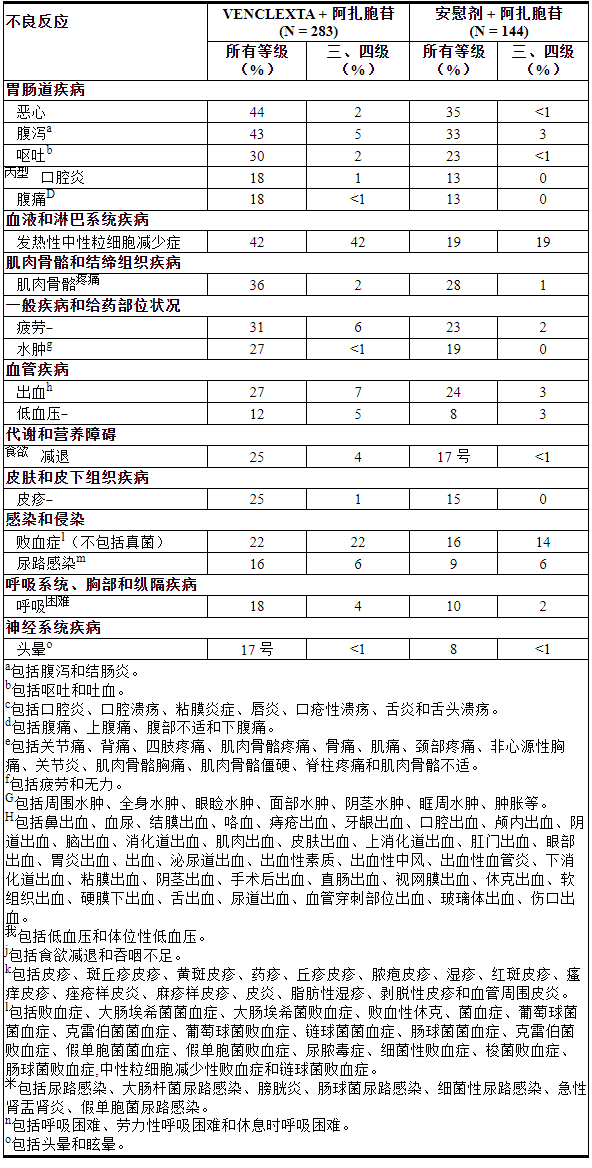
其他临床重要不良反应(所有等级)≥10%但不符合表 15 的标准或 <10% 如下:
肝胆疾病:胆囊炎/胆石症a (4%)
感染和侵染:b型肺炎(33%)
代谢和营养障碍:肿瘤溶解综合征(1%)
神经系统疾病:头痛c (11%)
调查:体重下降(13%)。
a包括急性胆囊炎、胆石症、胆囊炎和慢性胆囊炎。
b包括肺炎、肺部感染、真菌性肺炎、克雷伯菌肺炎、非典型肺炎、下呼吸道感染、病毒性肺炎、下呼吸道感染真菌、嗜血性肺炎、肺炎球菌性肺炎和呼吸道合胞病毒性肺炎。
c包括头痛和紧张性头痛。
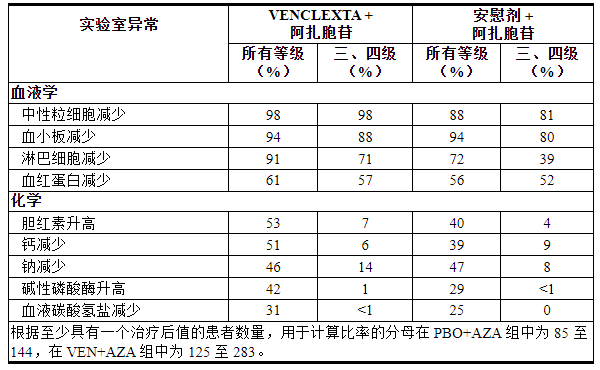
表 16列出了 VIALE-A 中发现的实验室异常情况。
表 16. 接受 VEN+AZA 治疗的 AML 患者中新出现或恶化的实验室异常 (≥10%),与 PBO+AZA 相比,所有级别的组间差异≥5%,3 级或 4 级反应的组间差异≥2%维亚莱-A
VENCLEXTA 与阿扎胞苷或地西他滨联用
M14-358 是一项针对新诊断 AML 患者的非随机试验,评估了 VENCLEXTA 与阿扎胞苷 (N=67) 或地西他滨 (N=13) 联用的安全性。基线时,患者年龄≥75岁,或患有基于以下标准之一而无法使用强化诱导化疗的合并症:基线ECOG表现状态为2-3,严重心脏或肺部合并症,中度肝功能损害,CLcr <45 mL/min,或其他合并症[参见临床研究(14.2)]。完成加速阶段后,患者接受 VENCLEXTA 400 mg 口服,每日一次,联合阿扎胞苷(75 mg/m 2,每个 28 天周期的第 1-7 天静脉内或皮下注射)或地西他滨(20 mg/m 2每个 28 天周期的第 1-5 天静脉注射)。
阿扎胞苷
当与阿扎胞苷联用时,VENCLEXTA暴露的中位持续时间为6.5个月(范围:0.1至38.1个月)。本试验中 VENCLEXTA 与阿扎胞苷联用的安全性与 VIALE-A 一致。
地西他滨
当与地西他滨联合给药时,VENCLEXTA 暴露的中位持续时间为 8.4 个月(范围:0.5 至 39 个月)。
接受 VENCLEXTA 联合地西他滨治疗的患者中,85% 报告出现严重不良反应,最常见(≥10%)为败血症(不包括真菌;46%)、发热性中性粒细胞减少症(38%)和肺炎(31%)。开始治疗后 30 天内发生了 1 例 (8%) 致命的菌血症不良反应。
38% 的患者因不良反应而永久停用 VENCLEXTA。导致永久停药的最常见不良反应(≥5%)是肺炎(8%)。
15% 的患者因不良反应而减少 VENCLEXTA 剂量。导致剂量减少(≥5%)的最常见不良反应是中性粒细胞减少症(15%)。
69% 的患者因不良反应而中断 VENCLEXTA 剂量。导致剂量中断的最常见不良反应(≥10%)是中性粒细胞减少症(38%)、发热性中性粒细胞减少症(23%)、白细胞减少症(15%)和肺炎(15%)。
最常见的不良反应(≥30%)是发热性中性粒细胞减少症(69%)、疲劳(62%)、便秘(62%)、肌肉骨骼疼痛(54%)、头晕(54%)、恶心(54%)、腹部不适疼痛(46%)、腹泻(46%)、肺炎(46%)、败血症(不包括真菌;46%)、咳嗽(38%)、发热(31%)、低血压(31%)、口咽痛(31%) )、水肿(31%)和呕吐(31%)。最常见的实验室异常(≥30%)为中性粒细胞减少(100%)、淋巴细胞减少(100%)、白细胞减少(100%)、血小板减少(92%)、钙减少(85%)、血红蛋白减少(69%)、葡萄糖增加(69%)、镁减少(54%)、钾减少(46%)、胆红素增加(46%)、白蛋白减少(38%)、碱性磷酸酶增加(38%)、钠减少(38%)、ALT 升高 (31%)、肌酐升高 (31%)、
VENCLEXTA 与低剂量阿糖胞苷联用
维亚莱-C
VIALE-C(一项双盲随机试验)评估了 VENCLEXTA 联合低剂量阿糖胞苷 (VEN+LDAC) (N=142) 与安慰剂联合低剂量阿糖胞苷 (PBO+LDAC) (N=68) 的安全性在新诊断的 AML 患者中进行的试验。基线时,患者年龄≥75岁,或患有基于以下标准之一而无法使用强化诱导化疗的合并症:基线ECOG表现状态为2-3,严重心脏或肺部合并症,中度肝功能损害,CLcr <45 mL/min,或其他合并症[参见临床研究(14.2)]。患者在完成 4 天的加速阶段后随机接受 VENCLEXTA 600 mg 口服,每日一次,联合低剂量阿糖胞苷(20 mg/m 2每个 28 天周期的第 1-10 天每天皮下注射一次)或安慰剂与低剂量阿糖胞苷组合。在接受 VEN+LDAC 的患者中,暴露于 VENCLEXTA 的中位持续时间为 3.9 个月(范围:<0.1 至 17.1 个月)。
接受 VEN+LDAC 治疗的患者中有 65% 报告出现严重不良反应,其中最常见(≥10%)的是肺炎(17%)、发热性中性粒细胞减少症(16%)和脓毒症(不包括真菌;12%)。接受 VEN+LDAC 治疗的患者中有 23% 发生致命不良反应,其中最常见(≥5%)的是肺炎(6%)和脓毒症(不包括真菌;7%)。
不良反应导致 25% 的患者永久停用 VENCLEXTA,9% 的患者减少剂量,63% 的患者中断剂量。导致VENCLEXTA永久停用的最常见不良反应(>2%)是肺炎(6%)。≥1%的患者需要减少剂量的不良反应为肺炎(1%)和血小板减少症(1%),≥5%的患者需要中断剂量的不良反应包括中性粒细胞减少症(20%)、血小板减少症(1%)和血小板减少症(1%)。 15%)、肺炎(8%)、发热性中性粒细胞减少症(6%)和败血症(不包括真菌;6%)。在实现白血病骨髓清除的患者中,32% 的患者因 ANC <500/微升而接受剂量中断。
表 17列出了 VIALE-C 中发现的不良反应。
表 17. VIALE-C 中接受 VEN+LDAC 治疗的 AML 患者的不良反应(≥10%),与 VIALE-C 中的 PBO+LDAC 相比,所有级别的组间差异≥5%,3 级或 4 级的组间差异≥2%
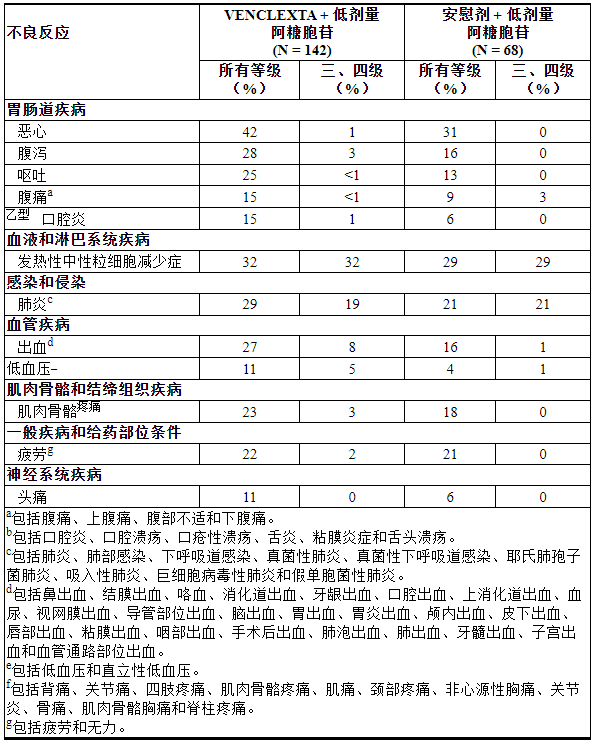
其他临床重要不良反应(所有等级)≥10%但不符合表 17的标准或<10%如下:
肝胆疾病:胆囊炎/胆石症a (1%)
感染和侵染:脓毒症b(不包括真菌;15%)、尿路感染c(8%)
代谢和营养障碍:食欲下降(19%)、肿瘤溶解综合征(6%)
神经系统疾病:头晕d (9%)
呼吸、胸部和纵隔疾病:呼吸困难e (10%)
调查:体重下降(9%)。
a包括胆囊炎和急性胆囊炎。
b包括败血症、菌血症、败血性休克、中性粒细胞减少性败血症、葡萄球菌菌血症、链球菌菌血症、细菌性败血症、埃希菌菌血症、假单胞菌菌血症和葡萄球菌败血症。
c包括尿路感染和大肠杆菌尿路感染。
d包括头晕和眩晕。
e包括呼吸困难和劳力性呼吸困难。
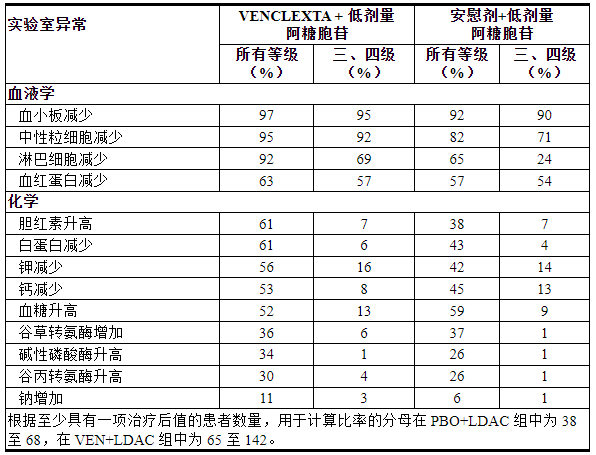
表 18描述了 VIALE-C 中发现的实验室异常情况。
表 18. VIALE 中接受 VEN+LDAC 治疗的 AML 患者中新出现或恶化的实验室异常 (≥10%),与 VIALE 中的 PBO+LDAC 相比,所有级别的组间差异≥5%,3 级或 4 级反应的组间差异≥2% -C
M14-387
VENCLEXTA 与低剂量阿糖胞苷联用(N=61)的安全性在 M14-387 中评价,M14-387 是一项新诊断 AML 患者的非随机、开放标签试验[见临床研究( 14.2 ) ]。基线时,患者年龄≥75岁,或患有基于以下至少一项标准而无法使用强化诱导化疗的合并症:基线ECOG表现状态为2-3,严重心脏或肺部合并症,中度肝功能损害、CLcr <45 mL/min,或其他合并症。完成加速阶段后,患者接受每日一次口服 VENCLEXTA 600 mg 联合低剂量阿糖胞苷(20mg/m 2每个 28 天周期的第 1-10 天皮下注射)。VENCLEXTA 与低剂量阿糖胞苷联合使用的安全性与 VIALE-C 一致。
7 药物相互作用
7.1 其他药物对 VENCLEXTA 的影响
强或中度 CYP3A 抑制剂或 P-gp 抑制剂
与强或中度CYP3A抑制剂或P-gp抑制剂同时使用增加venetoclax C max和AUC 0-INF [见临床药理学( 12.3 )],这可能增加VENCLEXTA毒性,包括TLS风险[见警告和注意事项( 5.1)]。
在 CLL/SLL 患者中,在开始时和加速阶段同时使用强 CYP3A 抑制剂是禁忌的[见禁忌症( 4 )]。
在有CLL/SLL患者服用稳定的每日剂量(在加速阶段后),考虑替代药物或调整VENCLEXTA剂量并更频繁地监测不良反应[见剂量和给药方法( 2.5,2.6 )]。
在有AML患者中,调整VENCLEXTA剂量并更频繁地监测不良反应[见剂量和给药方法( 2.5,2.6 )]。
终止抑制剂后2至3天恢复与强或中度CYP3A抑制剂或P-gp抑制剂同时使用之前使用的VENCLEXTA剂量[见剂量和给药方法( 2.5,2.6 ) ] 。
在 VENCLEXTA 治疗期间避免使用葡萄柚产品、塞维利亚橙子和杨桃,因为它们含有 CYP3A 抑制剂。
强或中度 CYP3A 诱导剂
与强CYP3A诱导剂同时使用降低venetoclax C max和AUC 0-INF [见临床药理学( 12.3 )],这可能降低VENCLEXTA疗效。避免 VENCLEXTA 与强 CYP3A 诱导剂或中度 CYP3A 诱导剂同时使用。
7.2 VENCLEXTA 对其他药物的影响
华法林
VENCLEXTA 的同时使用增加华法林 C max和 AUC 0-INF [见临床药理学( 12.3 )],这可能增加出血风险。对于同时使用华法林和 VENCLEXTA 的患者,更频繁地监测国际标准化比值 (INR)。
P-gp 底物
VENCLEXTA的同时使用增加P-gp底物的C max和AUC 0-INF [见临床药理学( 12.3 )],这可能增加这些底物的毒性。避免 VENCLEXTA 与 P-gp 底物同时使用。如果联合使用不可避免,请在 VENCLEXTA 之前至少 6 小时单独服用 P-gp 底物。
8 在特定人群中的使用
8.1 怀孕
风险总结
根据动物中的发现及其作用机制[见临床药理学( 12.1 )],当给予孕妇时VENCLEXTA可能会引起胚胎-胎儿伤害。尚无关于孕妇使用 VENCLEXTA 的可用数据来告知药物相关风险。根据 AUC,在器官形成期间对怀孕小鼠给予Venetoclax时,暴露量是人类推荐剂量 400 mg 每天暴露量的 1.2 倍时,具有胎儿毒性。告知孕妇对胎儿的潜在风险。
指定人群主要出生缺陷和流产的估计背景风险尚不清楚。所有怀孕都有出生缺陷、流产或其他不良后果的背景风险。在美国普通人群中,临床认可的妊娠中重大出生缺陷和流产的背景风险估计分别为 2% 至 4% 和 15% 至 20%。
数据
动物数据
在胚胎-胎儿发育研究中,在器官形成期间对怀孕小鼠和兔子施用维奈托克。在小鼠中, 150 mg/kg/天(母亲暴露量约为推荐剂量 400 mg 每天一次时人类暴露量的 1.2 倍), venetoclax与植入后丢失增加和胎儿体重下降相关。在小鼠或兔子中均未观察到致畸性。
8.2 哺乳期
风险总结
没有关于母乳中存在 VENCLEXTA 或对母乳喂养儿童或产奶量影响的数据。当给予哺乳大鼠时,维奈托克存在于乳汁中(参见数据)。
由于母乳喂养的儿童可能出现严重不良反应,建议女性在使用 VENCLEXTA 治疗期间以及最后一次给药后 1 周内不要母乳喂养。
数据
动物数据
产后 8 至 10 天给哺乳大鼠施用Venetoclax (单剂量;口服 150 mg/kg)。牛奶中的Venetoclax含量比血浆中低 1.6 倍。母体药物(venetoclax)占牛奶中药物相关物质总量的大部分,并含有微量的三种代谢物。
8.3 女性和男性的生殖潜力
当给予孕妇时VENCLEXTA可能致胎儿伤害[见在特殊人群中使用( 8.1 )]。
妊娠测试
在开始 VENCLEXTA 之前验证具有生殖潜力的女性的妊娠状态。
避孕
建议有生育潜力的女性在用 VENCLEXTA 治疗期间和最后一次给药后 30 天内使用有效的避孕措施。
不孕症
根据动物中的发现,VENCLEXTA 可能损害男性生育力[见非临床毒理学( 13.1 )]。
8.4 儿童使用
VENCLEXTA 在儿科患者中的安全性和有效性尚未确定。
幼年动物毒性数据
在一项幼年毒理学研究中,小鼠在 7 至 60 天龄期间以 10、30 或 100 mg/kg/天的剂量口服维奈托克。毒性的临床症状包括活动减少、脱水、皮肤苍白和驼背姿势(≥30 mg/kg/天)。此外,100 毫克/公斤/天时会出现死亡率和体重影响。其他与维奈托克相关的作用是≥10 mg/kg/天时淋巴细胞可逆性减少;对于20公斤体重的儿童,按mg/m 2计算,10毫克/公斤/天的剂量约为400毫克临床剂量的0.06倍。
8.5 老年人使用
慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
在 VENCLEXTA 单药治疗的 3 项开放标签试验中评估了 352 名既往接受过治疗的 CLL/SLL 患者的安全性,其中 57% (201/352) 年龄≥65 岁,18% (62/352) 年龄≥75 岁。
在联合治疗和单一治疗研究中,老年患者和年轻患者之间没有观察到安全性和有效性有临床意义的差异。
急性髓系白血病
在 VIALE-A 中接受 VENCLEXTA 联合阿扎胞苷治疗的 283 名患者中,96% 年龄≥65 岁,60% ≥75 岁。
在M14-358中接受VENCLEXTA联合地西他滨治疗的13名患者中,100%年龄≥65岁,62%年龄≥75岁。
在 VIALE-C 中接受 VENCLEXTA 联合低剂量阿糖胞苷治疗的 142 名患者中,92% 年龄≥65 岁,57% ≥75 岁。
VENCLEXTA 在 AML 患者中的临床研究没有包括足够数量的年轻人来确定 65 岁及以上患者的反应是否与年轻人不同。
8.6 肾损伤
由于 TLS 风险增加,肾功能降低的患者(CLcr <80 mL/min,通过 Cockcroft-Gault 公式计算)在开始用 VENCLEXTA 治疗时需要更强化的预防和监测以降低 TLS 风险[见剂量和给药方法] (2.1、2.2、2.3、2.4)和警告和注意事项(5.1 ) ] 。
对于有轻度、中度或重度肾受损(CLcr ≥15 mL/min)患者不建议调整剂量[见临床药理学( 12.3 )]。
8.7 肝损伤
对于轻度(Child-Pugh A)或中度(Child-Pugh B)肝功能不全的患者,不建议调整剂量。
对于严重肝受损患者(Child-Pugh C)减少VENCLEXTA的剂量;更频繁地监测这些患者的不良反应[见剂量和给药方法( 2.5,2.7 )和临床药理学( 12.3 )] 。
10 药物过量
VENCLEXTA 没有特效解毒剂。对于服药过量的患者,密切监测并提供适当的支持治疗;在加速阶段中断VENCLEXTA并仔细监测TLS的体征和症状以及其他毒性[见剂量和给药方法( 2.2,2.3,2.4,2.5 ) ] 。基于维奈托克的大量分布和广泛的蛋白质结合,透析不太可能导致维奈托克的显着去除。
11 描述
Venetoclax是一种 BCL-2 抑制剂。它是浅黄色至深黄色固体,经验式C 45 H 50 ClN 7 O 7 S,分子量为868.44。Venetoclax 的化学描述为 4-(4-{[2-(4-氯苯基)-4,4-二甲基环己-1-en-1-基]甲基}哌嗪-1-基)- N -({3-硝基) -4-[(四氢-2H-吡喃-4-基甲基)氨基]苯基}磺酰基)-2-(1H-吡咯并[2,3- b ]吡啶-5-基氧基)苯甲酰胺)并具有以下化学式结构:
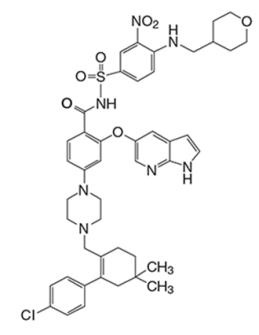
Venetoclax的水溶性非常低。
口服用 VENCLEXTA 片剂为淡黄色或米色片剂,含有 10、50 或 100 mg Venetoclax作为活性成分。每片还含有以下非活性成分:共聚维酮、胶体二氧化硅、聚山梨醇酯 80、硬脂酰富马酸钠和磷酸氢钙。此外,10毫克和100毫克包衣片剂还包括:氧化铁黄、聚乙烯醇、聚乙二醇、滑石粉和二氧化钛。50毫克包衣片剂还包含以下成分:氧化铁黄、氧化铁红、氧化铁黑、聚乙烯醇、滑石粉、聚乙二醇和二氧化钛。每片药片的一侧均凹有“V”字样,另一侧则凹有与药片强度相对应的“10”、“50”或“100”。
12 临床药理学
12.1 作用机制
Venetoclax是一种选择性的、口服生物可利用的 BCL-2(一种抗凋亡蛋白)小分子抑制剂。BCL-2 的过度表达已在 CLL 和 AML 细胞中得到证实,可介导肿瘤细胞存活并与化疗耐药相关。Venetoclax通过直接与 BCL-2 蛋白结合、取代 BIM 等促凋亡蛋白、触发线粒体外膜透化和半胱天冬酶激活,帮助恢复细胞凋亡过程。在非临床研究中,venetoclax已证明对过度表达 BCL-2 的肿瘤细胞具有细胞毒活性。
12.2 药效学
根据疗效的暴露反应分析,在 CLL/SLL 患者和 AML 患者的临床研究中观察到药物暴露与更大的反应可能性之间的关系。根据安全性暴露反应分析,在 AML 患者的临床研究中观察到药物暴露与某些安全事件发生可能性更大之间的关系。在 CLL/SLL 患者中,单药治疗剂量高达 1200 mg 以及与利妥昔单抗联合治疗剂量高达 600 mg 时,未观察到暴露-安全性关系。
心脏电生理学
在一项开放标签、单臂试验中,对 176 例既往接受过血液系统恶性肿瘤治疗的患者进行了评估,多次剂量 VENCLEXTA 至 1200 mg 每日一次(最大批准推荐剂量的 2 倍)对 QTc 间期的影响。VENCLEXTA 对 QTc 间期无大影响(即 >20 ms), venetoclax暴露与 QTc 间期变化之间没有关系。
12.3 药代动力学
每日一次400 mg与低脂膳食给药后,Venetoclax平均(±标准差)稳态C max为2.1 ± 1.1 mcg/mL,AUC 0-24h为32.8 ± 16.9 mcg•h/mL。Venetoclax稳态 AUC 在 150 至 800 mg 剂量范围内按比例增加(最大批准推荐剂量的 0.25 至 1.33 倍)。Venetoclax的药代动力学不随时间而改变。
吸收
在进食条件下多次口服给药后 5 至 8 小时达到Venetoclax的最大血浆浓度。
食物的影响
低脂膳食(约 512 卡路里,25% 脂肪热量,60% 碳水化合物热量和 15% 蛋白质热量)给药使Venetoclax暴露量增加约 3.4 倍,与高脂肪膳食(约 753 卡路里,55 % 脂肪热量、28% 碳水化合物热量和 17% 蛋白质热量)与禁食条件相比,维奈托克暴露量增加了 5.1 至 5.3 倍。
分配
Venetoclax与人血浆蛋白高度结合,在 1-30 微摩尔 (0.87-26 mcg/mL) 浓度范围内,血浆中未结合分数 <0.01。平均血液与血浆比率为0.57。患者中Venetoclax的表观分布容积 (Vd ss /F)范围为 256-321 L。
消除
Venetoclax的终末消除半衰期约为 26 小时。
代谢
Venetoclax在体外主要由 CYP3A 代谢。血浆中鉴定的主要代谢物 M27 对 BCL-2 的抑制活性在体外比Venetoclax 至少低 58 倍,其 AUC 为母体 AUC 的 80%。
排泄
健康受试者单次口服放射性标记的[ 14 C] -venetoclax 200 mg后,9天内在粪便中回收了>99.9%的剂量(21%为未改变),在尿液中回收了<0.1%。
特定人群
根据年龄(19至93岁)、性别、体重、轻度至重度肾功能不全(CLcr 15至89 mL/min,通过Cockcroft-Gault计算)或轻度至中度,未观察到维奈托克的药代动力学存在临床显着差异肝功能损害(总胆红素和天冬氨酸转氨酶 (AST) 正常 > 正常上限 (ULN) 或总胆红素为 ULN 的 1 至 3 倍)。终末期肾病(CLcr <15 mL/min)或透析对维奈托克药代动力学的影响尚不清楚。
种族或族裔群体
在美国入组的白人、黑人和亚洲患者中,未观察到Venetoclax的药代动力学存在临床显着差异。在 771 名 AML 患者中,来自亚洲国家 [中国 (5.6%)、日本 (5.5%)、韩国 (2.1%) 和台湾 (0.9%)] 的亚洲患者的维奈托克暴露量比非亚洲人群高 63 %。
肝损伤患者
单剂量VENCLEXTA 50 mg后,有严重肝受损受试者(Child-Pugh C)与有正常肝功能受试者比较venetoclax全身暴露(AUC 0-INF )是2.7倍高[见剂量和给药方法( 2.7 )和在特定人群中的使用(8.7)]。轻度或中度肝受损受试者与肝功能正常受试者之间未观察到维奈托克全身暴露的临床相关差异。
药物相互作用研究
临床研究
当与阿扎胞苷、阿奇霉素、阿糖胞苷、地西他滨、胃酸减少剂、奥比妥珠单抗或利妥昔单抗共同给药时,未观察到维奈托克药代动力学存在临床显着差异。
酮康唑
同时使用酮康唑(一种强CYP3A、P-gp和BCRP抑制剂)400 mg每天一次共7天增加venetoclax C max 130%和AUC 0-INF 540% [见药物相互作用( 7.1 )]。
利托那韦
同时使用利托那韦(一种强CYP3A、P-gp和OATP1B1/B3抑制剂)50 mg每天一次共14天增加venetoclax C max 140%和AUC 690% [见药物相互作用( 7.1 )]。
泊沙康唑
泊沙康唑(一种强效 CYP3A 和 P-gp 抑制剂)300 mg 与 VENCLEXTA 50 mg 和 100 mg 联合使用 7 天,与单独给予 VENCLEXTA 400 mg 相比,venetoclax C max分别升高 61% 和 86%。Venetoclax AUC 0-24h分别较高90%和144% [见药物相互作用( 7.1 ) ]。
利福平
同时使用单剂量利福平 (一种 OATP1B1/1B3 和 P-gp 抑制剂)600 mg 可使Venetoclax C max增加106%,AUC 0-INF增加 78%。同时使用多剂量利福平(作为强CYP3A诱导剂)600 mg每天一次共13天减低venetoclax C max 42%和AUC 0-INF 71% [见药物相互作用( 7.1 )]。
华法林
单次400 mg剂量VENCLEXTA与5 mg华法林的同时使用导致R-华法林和S-华法林的C max和AUC 0-INF增加18%至28% [见药物相互作用( 7.2 )]。
地高辛
单剂量VENCLEXTA 100 mg与地高辛(一种P-gp底物)0.5 mg同时使用增加地高辛C max 35%和AUC 0-INF 9% [见药物相互作用( 7.2 )]。
体外研究
Venetoclax不是 CYP1A2、CYP2B6、CYP2C19、CYP2D6 或 CYP3A4 的抑制剂或诱导剂。Venetoclax是 CYP2C8、CYP2C9 和 UGT1A1 的弱抑制剂。
Venetoclax不是 UGT1A4、UGT1A6、UGT1A9 或 UGT2B7 的抑制剂。
Venetoclax是 P-gp 和 BCRP 的抑制剂和底物以及 OATP1B1 的弱抑制剂。
Venetoclax不是 OATP1B3、OCT1、OCT2、OAT1、OAT3、MATE1 或 MATE2K 的抑制剂。
13 非临床毒理学
13.1 致癌、突变、生育能力受损
在一项为期 6 个月的转基因 (Tg.rasH2) 小鼠研究中,口服剂量高达 400 mg/kg/天的Venetoclax和单次口服剂量水平 250 mg/kg 时,Venetoclax和 M27(一种主要的人类代谢物)均不致癌。千克/天 M27。
Venetoclax在体外细菌致突变性 (Ames) 测定中不致突变,在使用人外周血淋巴细胞的体外染色体畸变测定中不诱导数值或结构畸变,并且在体内小鼠骨髓微核测定中在一定剂量下不致断裂。高达 835 毫克/公斤。在体外 Ames 和染色体畸变试验中,M27 代谢物的基因毒性活性呈阴性。
在雄性和雌性小鼠中进行了生育力和早期胚胎发育研究。这些研究通过植入评估交配、受精和胚胎发育。在剂量高达 600 mg/kg/天时,venetoclax对发情周期、交配、生育力、黄体、子宫植入或每窝活胚胎没有影响。然而,根据在狗中观察到的睾丸毒性(生殖细胞损失),在暴露量低至人类 AUC 暴露量 400 mg 的 0.5 倍时,存在对人类男性生育能力的风险。
13.2 动物毒理学和/或药理学
在狗身上,venetoclax引起多种组织的单细胞坏死,包括胆囊、外分泌胰腺和胃,但没有证据表明组织完整性遭到破坏或器官功能障碍。这些发现的程度从最小到轻微。经过 4 周的给药期和随后的 4 周恢复期后,一些组织中仍然存在最小程度的单细胞坏死,并且在较长时间的给药或恢复期后尚未评估可逆性。
此外,在狗每天服用约 3 个月后,由于黑色素的流失, venetoclax导致毛发逐渐变白。
14 临床研究
14.1 慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
与奥比妥珠单抗联用
CLL14 (BO25323) 是一项随机 (1:1)、多中心、开放标签、主动对照试验 (NCT02242942),评估 VENCLEXTA 联合 obinutuzumab (VEN+G) 与 obinutuzumab 联合苯丁酸氮芥 (GClb) 的疗效和安全性) 对于既往未接受治疗且合并有医疗状况的 CLL 患者(总累积疾病评定量表 [CIRS] 评分 >6 或 CLcr <70 mL/min)。该试验要求肝转氨酶和总胆红素≤正常上限的2倍,并排除有里氏转化或除眼、耳、鼻、喉器官系统外CIRS评分为4的任何个体器官/系统损伤的患者。
所有患者均在第 1 天、第 1 周期的第 8 和第 15 天以及每个后续周期的第 1 天接受 obinutuzumab 1000 mg(第一剂可分为第 1 天和第 2 天分别为 100 mg 和 900 mg),总共 6 次循环。VEN+G 组中的患者在第 1 周期第 22 天开始 VENCLEXTA 5 周加速给药方案[见剂量和给药方法 ( 2.2 , 2.4 )],并从第 3 周期第 1 天开始每日一次口服 VENCLEXTA 400 mg,直到第 12 周期的最后一天。随机分配至 GC1b 组的患者在第 1 至 12 周期的第 1 天和第 15 天口服苯丁酸氮芥 0.5 mg/kg。每个周期为 28 天。
共有 432 名患者被随机分配,每组 216 名。各组之间的基线人口统计学和疾病特征相似。中位年龄为 72 岁(范围:41 至 89 岁),其中 89% 为白人,67% 为男性;36% 和 43% 分别为 Binet B 期和 C 期,88% 的东部肿瘤合作组 (ECOG) 表现状态 <2。CIRS 评分中位数为 8.0(范围:0 至 28),58% 的患者 CLcr <70 mL/min。8% 的患者检测到 17p 缺失, 10% 的患者检测到TP53突变,19% 的患者检测到 11q 缺失,57% 的患者检测到未突变的IgVH。
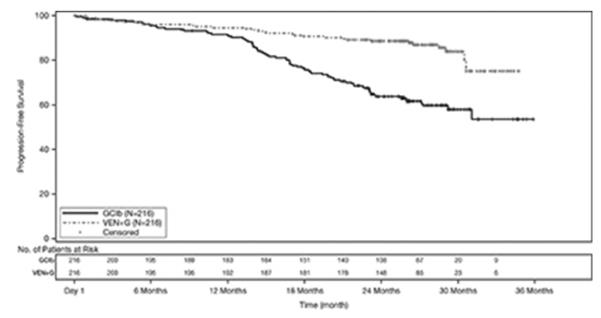
疗效基于独立审查委员会 (IRC) 评估的无进展生存期 (PFS)。PFS 随访的中位持续时间为 28 个月(范围:0 至 36 个月)。CLL14的功效结果示于表19中。PFS 的 Kaplan-Meier 曲线如图 1所示。
表 19. CLL14 的疗效结果
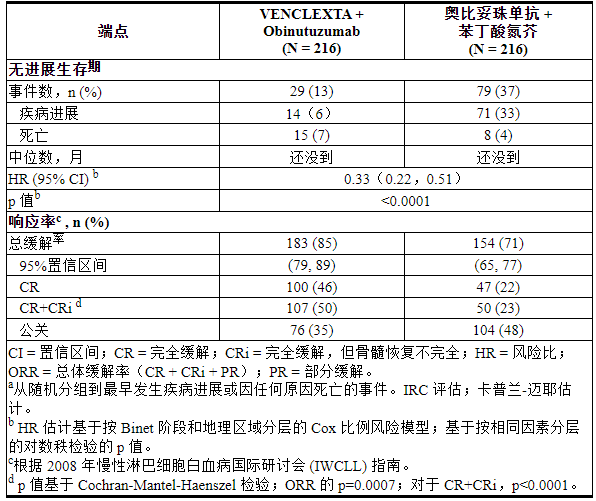
图 1. IRC 评估的 CLL14 无进展生存期的 Kaplan-Meier 曲线
在分析时,尚未达到中位总生存期 (OS),只有不到 10% 的患者经历过事件。OS 的中位随访时间为 28 个月。
使用等位基因特异性寡核苷酸聚合酶链反应(ASO-PCR)评估微小残留病(MRD)。阴性状态的定义是每 10 4 个白细胞中少于 1 个 CLL 细胞。表20显示了治疗完成后3个月的MRD阴性率,无论反应如何,以及达到CR的患者。在此评估中,VEN+G 组中 134 名外周血 MRD 阴性的患者的骨髓标本相匹配;其中,122 名患者 (91%) 外周血和骨髓 MRD 均为阴性。
表 20. CLL14 治疗完成后三个月的最小残留疾病阴性率
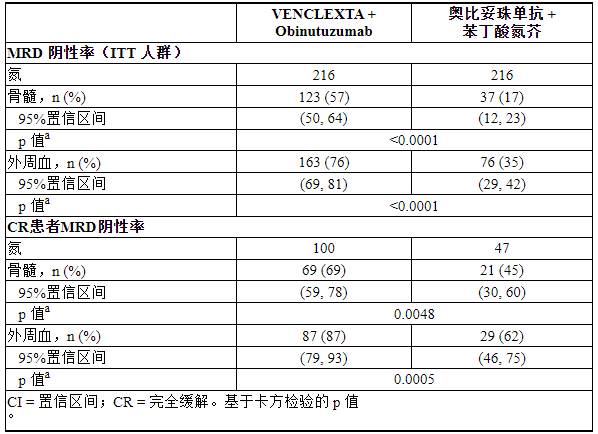
治疗完成后12个月,用VEN+G治疗的患者外周血MRD阴性率为58%(126/216),用GClb治疗的患者为9%(20/216)。
与利妥昔单抗联合使用
MURANO 是一项随机 (1:1)、多中心、开放标签试验 (NCT02005471),评估 VENCLEXTA 联合利妥昔单抗 (VEN+R) 与苯达莫司汀联合利妥昔单抗 (B+R) 在患有以下疾病的患者中的疗效和安全性: CLL 之前至少接受过一种治疗。VEN+R 组中的患者完成了 VENCLEXTA 5 周递增给药方案[见剂量和给药方法( 2.2,2.4 )] ,并在没有利妥昔单抗的情况下从第 1 周期第 1 天开始口服 VENCLEXTA 400 mg 每日一次,持续 24 个月疾病进展或不可接受的毒性。在第 1 周期第 1 天静脉注射剂量为 375 mg/m 2和 500 mg/m 2的 5 周剂量增加后开始使用利妥昔单抗在第 2-6 个周期的第 1 天静脉注射。随机分配至B+R的患者在第1天和第2天静脉注射70 mg/m 2苯达莫司汀,持续6个周期,并按上述剂量和时间表联合利妥昔单抗。每个周期为28天。
共有 389 名患者被随机分配:194 名患者接受 VEN+R 组,195 名患者接受 B+R 组。VEN+R 组和 B+R 组之间的基线人口统计和疾病特征相似。中位年龄为 65 岁(范围:22 至 85 岁),97% 为白人,74% 为男性,99% 的 ECOG 体力状态 <2。既往治疗中位数为 1(范围:1 至 5);59% 之前接受过 1 种治疗,26% 之前接受过 2 种治疗,16% 之前接受过 3 种或更多治疗。先前的治疗包括烷化剂(94%)、抗CD20抗体(77%)、B细胞受体通路抑制剂(2%)和先前的嘌呤类似物(81%,包括55%的氟达拉滨/环磷酰胺/利妥昔单抗)。24% 的患者检测到 17p 缺失, 25% 的患者检测到TP53突变,32% 的患者检测到 11q 缺失,IgVH未突变占 63%。
疗效基于 IRC 评估的 PFS。PFS 的中位随访时间为 23.4 个月(范围:0 至 37.4+ 个月)。MURANO的功效结果显示在表21中。PFS 的 Kaplan-Meier 曲线如图 2所示。
表 21. MURANO 中 IRC 评估的疗效结果
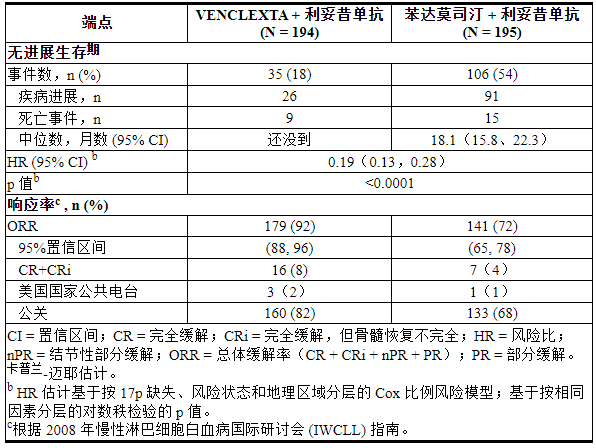
图 2. IRC 评估的 MURANO 无进展生存期的 Kaplan-Meier 曲线
最后一剂利妥昔单抗后 3 个月,达到 PR 或更好的患者外周血 MRD 阴性率在 VEN+R 组中为 54% (104/194),在 B+ 组中为 12% (23/195) R臂。此时 VEN+R 组中 MRD 阴性 CR/CRi 率为 3% (6/194),B+R 组中为 2% (3/195)。
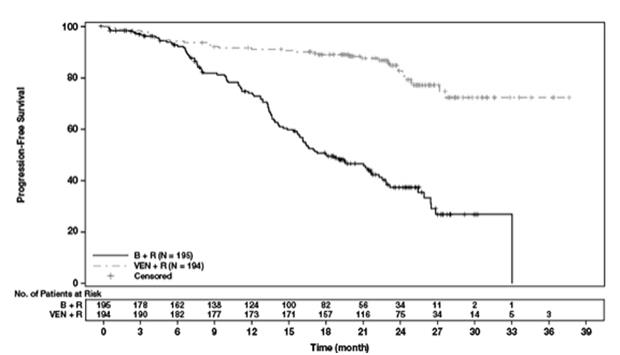
71 个月随访
经过 71 个月的总体随访,研究者评估 VEN+R 组的中位 PFS 为 53.6 个月(95% CI:48.4,57.0),B+ 组为 17.0 个月(95% CI:15.5,21.7) R臂。两组均未达到中位 OS。VEN+R 组中有 16% (32/194) 的患者死亡,B+R 组中有 33% (64/195) 的患者死亡(分层 HR 0.40;95% CI [0.26, 0.62])。OS 的 Kaplan-Meier 曲线如图 3所示。
图 3. MURANO 总体生存率的 Kaplan-Meier 曲线
单一疗法
VENCLEXTA 单药疗法对既往治疗过的 CLL 或 SLL 的疗效基于三项单臂试验。
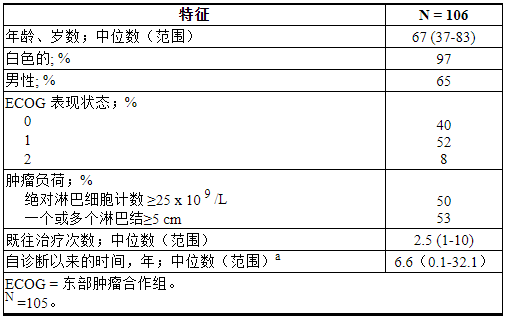
M13-982
M13-982 (NCT01889186) 是一项开放标签、多中心试验,入组了 106 名患有 17p 缺失的 CLL 患者,这些患者之前至少接受过一种治疗。在该试验中,使用 Vysis CLL FISH 探针试剂盒(经 FDA 批准用于选择接受 VENCLEXTA 治疗的患者)的患者外周血标本中确认了 17p 缺失。完成剂量递增方案后患者接受VENCLEXTA 400 mg口服每天1次[见剂量和给药方法( 2.2,2.4 )]。
疗效基于 IRC 评估的总体缓解率 (ORR)。
表 22总结了试验人群的基线人口统计和疾病特征。
表 22. M13-982 中的基线患者特征
评估时的中位治疗时间为 12.1 个月(范围:0 至 21.5 个月)。功效结果示于表23中。
表 23. 对于先前治疗过 M13-982 中 17p 缺失的 CLL 患者,每个 IRC 的疗效结果
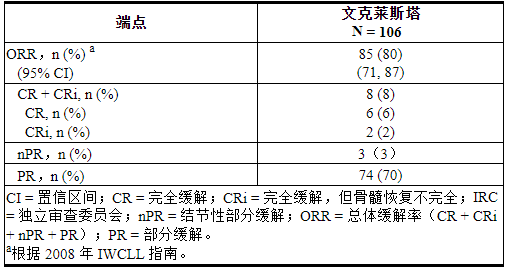
首次反应的中位时间为 0.8 个月(范围:0.1 至 8.1 个月)。
根据后来的数据截止日期和研究者评估的疗效,缓解持续时间 (DOR) 范围为 2.9 至 32.8+ 个月。中位随访 22 个月后,尚未达到中位 DOR。
对于用 VENCLEXTA 治疗后达到 CR 或 CRi 的患者,评估外周血和骨髓中的微小残留病。百分之三 (3/106) 的外周血和骨髓中 MRD 呈阴性(每 10 4 个白细胞少于 1 个 CLL 细胞)。
M12-175
M12-175 (NCT01328626) 是一项开放标签、多中心试验,入组了既往接受过治疗的 CLL 或 SLL 患者,包括 17p 缺失患者。对 67 名患者(59 名 CLL,8 名 SLL)进行了疗效评估,这些患者在完成剂量递增方案后每天口服一次 VENCLEXTA 400 mg。患者持续使用该剂量直至疾病进展或出现不可接受的毒性。评估时的中位治疗持续时间为 22.1 个月(范围:0.5 至 71.7 个月)。
中位年龄为 65 岁(范围:42 至 84 岁),其中 78% 为男性,87% 为白人。先前治疗的中位数为 3 次(范围:1 至 11)。基线时,67% 的患者有一个或多个淋巴结≥5 cm,30% 的患者有 ALC ≥25 x 10 9 /L,33% 记录有未突变的IgVH,21% 记录有 17p 缺失。
功效基于 2008 年 IWCLL 指南并由 IRC 评估。ORR 为 76%(95% CI:64%、86%),CR + CRi 率为 10%,PR 率为 66%。中位 DOR 为 36.2 个月(范围:2.4 至 52.4 个月)。
M14-032
M14-032 (NCT02141282) 是一项开放标签、多中心试验,入组之前曾接受过 ibrutinib 或 idelalisib 治疗并在治疗后病情进展的 CLL 患者。完成剂量递增方案后患者接受VENCLEXTA 400 mg口服每天1次[见剂量和给药方法( 2.2,2.4 )]。患者持续使用该剂量直至疾病进展或出现不可接受的毒性。截至分析时,中位治疗持续时间为 19.5 个月(范围:0.1 至 39.5 个月)。
在 127 名接受治疗的患者中(91 名既往接受过 ibrutinib,36 名接受过 idelalisib),中位年龄为 66 岁(范围:28 至 85 岁),70% 为男性,92% 为白人。先前治疗的中位数为 4 次(范围:1 至 15)。基线时,41% 的患者有一个或多个淋巴结≥5 cm,31% 的患者 ALC ≥25 x 10 9 /L,57% 记录有未突变的IgVH,39% 记录有 17p 缺失。
功效基于 2008 年 IWCLL 指南,并由 IRC 进行评估。ORR 为 70%(95% CI:61%、78%),CR + CRi 率为 5%,PR 率为 65%。中位随访时间为 19.9 个月(范围:2.9 至 36 个月),但未达到中位 DOR。
14.2 急性髓系白血病
VENCLEXTA 在新诊断 AML 成年患者中进行研究,这些患者年龄为 75 岁或以上,或患有基于以下标准之一而无法使用强化诱导化疗的合并症:基线 ECOG 表现状态为 2-3、严重心脏病或肺部合并症、中度肝功能损害、CLcr <45 mL/min 或其他合并症。
与阿扎胞苷或地西他滨联用
VIALE-A 是一项随机 (2:1)、双盲、安慰剂对照、多中心试验 (NCT02993523),评估 VENCLEXTA 联合阿扎胞苷 (VEN+AZA) 与安慰剂联合阿扎胞苷 (PBO+AZA) 的疗效和安全性)。
患者完成加速给药方案后第1-28天口服VENCLEXTA 400 mg每天一次[见剂量和给药方法( 2.3 )]或安慰剂联合阿扎胞苷75 mg/m 2在第1天静脉内或皮下注射每个 28 天周期的 7 个周期从第 1 周期第 1 天开始。在启动期间,患者接受 TLS 预防并住院进行监测。
一旦骨髓评估证实缓解(定义为第1周期治疗后白血病原始细胞少于5%并伴有血细胞减少),VENCLEXTA或安慰剂中断长达14天或直到ANC≥500/微升和血小板计数≥50 × 10 3 /微升。对于第 1 周期结束时患有耐药性疾病的患者,在第 2 或 3 周期后并根据临床指示进行骨髓评估。中断后,阿扎胞苷与 VENCLEXTA 或安慰剂在同一天恢复。在治疗血液学毒性的临床试验中实施阿扎胞苷剂量减少[见剂量和给药方法( 2.5 )]。患者继续治疗直至疾病进展或出现不可接受的毒性。
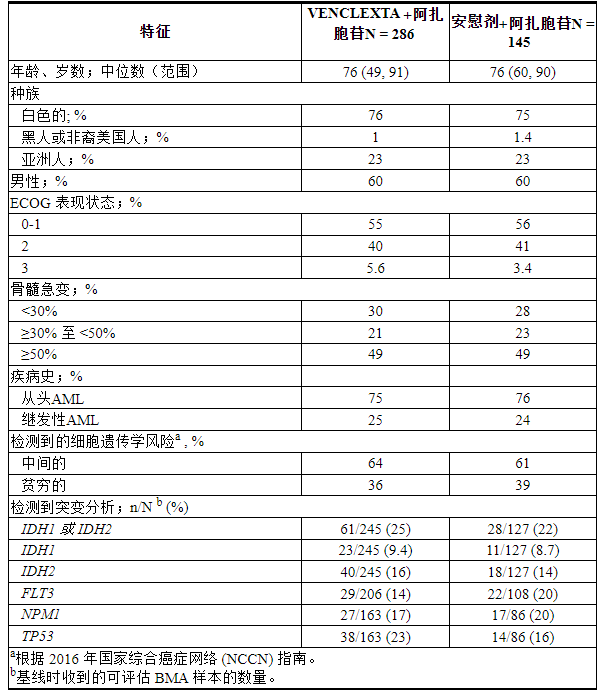
共有 431 名患者被随机分配:286 名患者接受 VEN+AZA 组,145 名患者接受 PBO+AZA 组。基线人口统计和疾病特征如表24所示。
表 24. VIALE-A 中 AML 患者的基线人口统计和疾病特征
疗效基于总生存期(OS),从随机分组之日起一直到全因死亡为止。VEN+AZA 组合的 OS 优于 PBO+AZA。
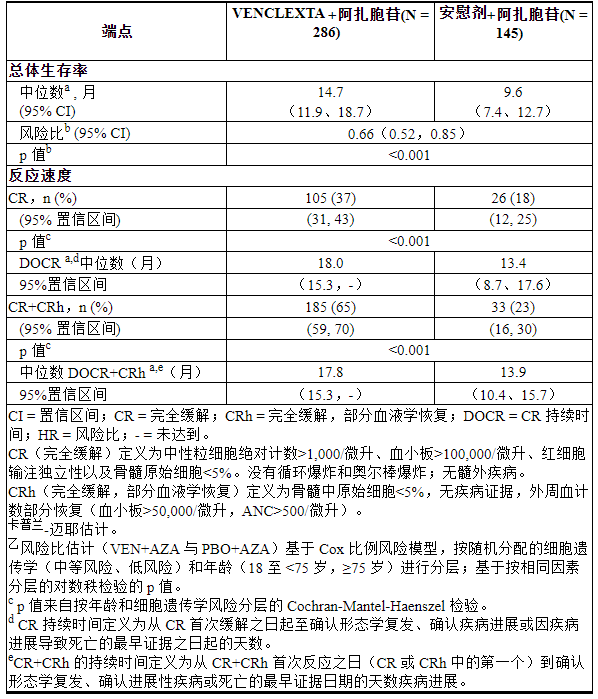
OS的Kaplan-Meier曲线如图4所示。VIALE-A的功效结果如表25所示。
图4 . VIALE-A 中总体生存的 Kaplan-Meier 曲线
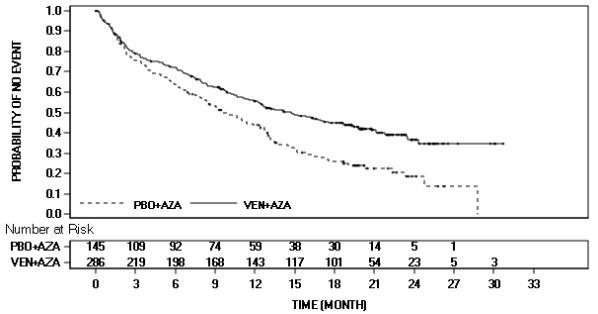
表 25. VIALE-A 中的功效结果
在接受 VEN+AZA 治疗的患者中,155 名患者在基线时依赖红细胞 (RBC) 和/或血小板输注;在这些患者中,49% (76/155) 在任何连续 ≥56 天的基线后期间不再依赖红细胞和血小板输注。在接受 VEN+AZA 治疗的患者中,131 例在基线时不依赖红细胞和血小板输注,69% (90/131) 在任何连续≥56 天的基线后期间保持不依赖输血。在接受 PBO+AZA 治疗的患者中,81 名患者在基线时依赖红细胞和/或血小板输注;在这些患者中,27% (22/81) 的患者在任何连续 ≥56 天的基线后期间不再依赖红细胞和血小板输注。在接受 PBO+AZA 治疗的患者中,64 例在基线时独立于红细胞和血小板输注,
VEN+AZA 治疗中首次出现 CR 或 CRh 反应的中位时间为 1.0 个月(范围:0.6 至 14.3 个月)。
M14-358
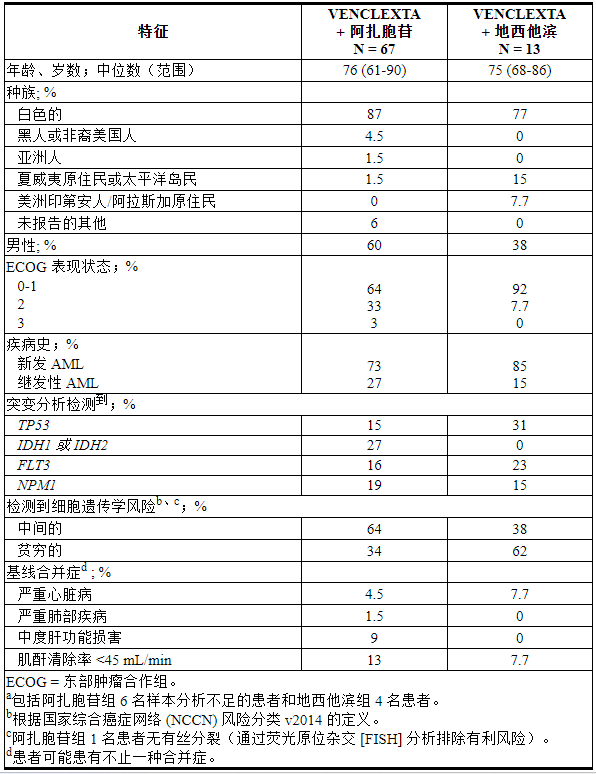
M14-358 (NCT02203773) 是一项非随机、开放标签试验,评估 VENCLEXTA 联合阿扎胞苷 (N=84) 或地西他滨 (N=31) 对新诊断 AML 患者的疗效。在这些患者中,67 名接受阿扎胞苷联合治疗的患者和 13 名接受地西他滨联合治疗的患者年龄为 75 岁或以上,或患有无法使用强化诱导化疗的合并症。
完成加速给药方案后,患者接受VENCLEXTA 400 mg口服每日一次[见剂量和给药方法( 2.3 )]联合阿扎胞苷(75 mg/m 2静脉内或皮下注射,每28天的第1-7天)周期从第 1 周期第 1 天开始)或地西他滨(20 mg/m 2从第 1 周期开始的每个 28 天周期的第 1-5 天静脉注射(第 1 天)。在启动阶段,患者接受 TLS 预防并住院监测。患者继续治疗直至疾病进展或出现不可接受的毒性。一旦骨髓评估证实缓解(定义为白血病原始细胞低于 5%,且第 1 周期治疗后出现血细胞减少),VENCLEXTA 中断长达 14 天,或直至 ANC ≥500/微升且血小板计数≥50 × 10 3 /微升。在治疗血液毒性的临床试验中实施阿扎胞苷剂量减少[见剂量和给药方法( 2.5 )]。临床试验中未实施地西他滨剂量减少。基线人口统计和疾病特征如表26所示。
表 26. 用 VENCLEXTA 联合阿扎胞苷或地西他滨治疗的 AML 患者的基线患者特征
功效结果如表27所示。
表 27. 用 VENCLEXTA 联合阿扎胞苷或地西他滨治疗的新诊断 AML 患者的疗效结果
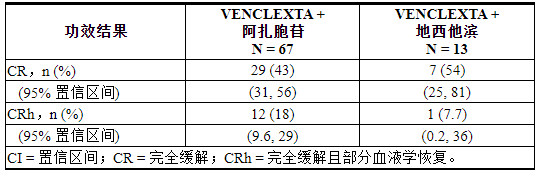
VENCLEXTA 联合阿扎胞苷的中位随访时间为 15.9 个月(范围:0.4 至 40.3 个月)。CR 的中位持续时间为 23.8 个月(95% CI:15.4,-),CR+CRh 的中位持续时间为 26.5 个月(95% CI:17.4,-)。
VENCLEXTA 与地西他滨联合治疗的中位随访时间为 11.0 个月(范围:0.7 至 38.8 个月)。CR 的中位持续时间为 12.7 个月(95% CI:1.4,-),CR+CRh 的中位持续时间为 12.7 个月(95% CI:1.4,20.0)。
CR 持续时间定义为从首次记录 CR 到首次复发、临床疾病进展或因疾病进展死亡(以最早发生者为准)的时间。CR+CRh 的持续时间定义为从首次记录 CR 或 CRh 到首次复发、临床疾病进展或因疾病进展死亡(以最早发生者为准)的时间。
用 VENCLEXTA 联合阿扎胞苷治疗患者至首次 CR 或 CRh 的中位时间为 1.0 个月(范围:0.7 至 8.9 个月)。
用 VENCLEXTA 联合地西他滨治疗患者达到首次 CR 或 CRh 的中位时间为 1.9 个月(范围:0.8 至 4.2 个月)。
在接受 VENCLEXTA 联合阿扎胞苷治疗的患者中,12% (8/67) 随后接受了干细胞移植。
该试验招募了另外 35 名患者(年龄范围:65 至 74 岁),这些患者没有已知的合并症而无法使用强化诱导化疗,并接受 VENCLEXTA 联合阿扎胞苷 (N=17) 或地西他滨 (N=18) 联合治疗。
对于 VENCLEXTA 联合阿扎胞苷治疗的 17 例患者,CR 率为 35%(95% CI:14%、62%)。CRh 率为 41%(95% CI:18%、67%)。九名(53%)患者随后接受了干细胞移植。
对于 VENCLEXTA 联合地西他滨治疗的 18 例患者,CR 率为 56%(95% CI:31%、79%)。CRh 率为 22%(95% CI:6.4%、48%)。四名(22%)患者随后接受了干细胞移植。
与低剂量阿糖胞苷联合使用
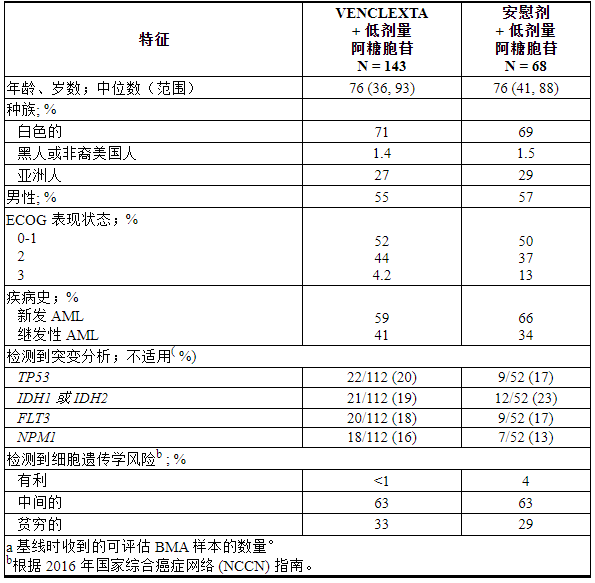
VIALE-C 是一项随机 (2:1)、双盲、安慰剂对照、多中心试验 (NCT03069352),评估 VENCLEXTA 联合低剂量阿糖胞苷 (VEN+LDAC) 与安慰剂联合低剂量阿糖胞苷的疗效和安全性。剂量阿糖胞苷(PBO+LDAC)。
患者完成加速给药方案后第1-28天口服VENCLEXTA 600 mg每天一次[见剂量和给药方法( 2.3 )]或安慰剂联合阿糖胞苷20 mg/m 2皮下注射第1-10天每天一次从周期 1 第 1 天开始的每个 28 天周期的周期。在启动阶段,患者接受 TLS 预防并住院进行监测。
一旦骨髓评估证实缓解(定义为第1周期治疗后白血病原始细胞少于5%并伴有血细胞减少),VENCLEXTA或安慰剂中断长达14天或直到ANC≥500/微升和血小板计数≥50 × 10 3 /微升。对于第 1 周期结束时患有耐药性疾病的患者,在第 2 或 3 周期后并根据临床指示进行骨髓评估。中断后,LDAC 与 VENCLEXTA 或安慰剂在同一天恢复。患者继续接受治疗,直到疾病进展或出现不可接受的毒性。基线人口统计和疾病特征如表28所示。
表 28. VIALE-C 中 AML 患者的基线人口统计和疾病特征
疗效基于 CR 发生率和 CR 持续时间,以及 CR+CRh 发生率、CR+CRh 持续时间以及从输血依赖到输血独立的转化率的支持证据。VEN+LDAC 组的 CR 率为 27%(95% CI:20%、35%),CR 中位持续时间为 11.1 个月(95% CI:6.1,-),PBO+ 组的 CR 率为LDAC 组的率为 7.4% (95% CI: 2.4%, 16%),中位 CR 持续时间为 8.3 个月 (95% CI: 3.1, - )。VEN+LDAC 组的 CR+CRh 率为 47%(95% CI:39%、55%),PBO+LDAC 组的 CR+CRh 率为 15%(95% CI:7.3%、25%),中位持续时间VEN+LDAC 治疗的 CR+CRh 为 11.1 个月,PBO+LDAC 治疗的 CR+CRh 为 6.2 个月。VEN+LDAC 治疗中首次出现 CR 或 CRh 反应的中位时间为 1.0 个月(范围:0.7 至 5.8 个月)。
在接受 VEN+LDAC 治疗的患者中,有 111 名患者在基线时依赖红细胞和/或血小板输注;在这些患者中,33% (37/111) 的患者在任何连续 ≥56 天的基线后期间不再依赖红细胞和血小板输注。在接受 VEN+LDAC 治疗的患者中,32 例在基线时不依赖红细胞和血小板输注,50% (16/32) 在任何连续≥56 天的基线后期间保持不依赖输血。
在接受 PBO+LDAC 治疗的患者中,有 55 名患者在基线时依赖红细胞和/或血小板输注;在这些患者中,13% (7/55) 的患者在任何连续 ≥56 天的基线后期间不再依赖红细胞和血小板输注。在接受 PBO+LDAC 治疗的患者中,13 例在基线时不依赖红细胞和血小板输注,31% (4/13) 在任何连续≥56 天的基线后期间保持不依赖输血。
与 PBO+LDAC 相比,VEN+LDAC 并未显着改善 OS。OS 的风险比 (HR) 为 0.75 (95% CI: 0.52, 1.07);p 值 0.114。VEN+LDAC 组的中位 OS 为 7.2 个月(95% CI:5.6,10.1),PBO+LDAC 组的中位 OS 为 4.1 个月(95% CI:3.1,8.8)。
M14-387
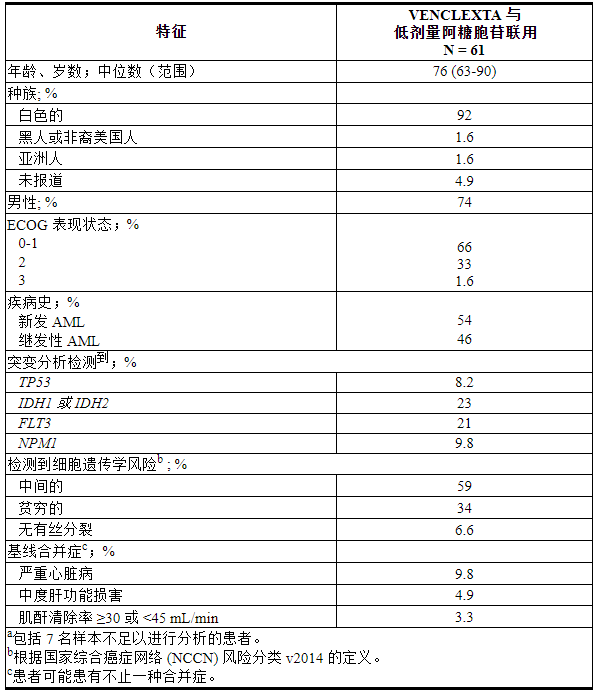
M14-387 (NCT02287233) 是一项非随机、开放标签试验,评估 VEN+LDAC (N=82) 对新诊断的 AML 患者的疗效,包括之前因血液疾病而使用过低甲基化药物的患者。这些患者中,61 名年龄在 75 岁或以上,或患有合并症而无法使用强化诱导化疗。
患者在完成加速阶段后第1-28天接受VENCLEXTA 600 mg口服每天一次[见剂量和给药方法( 2.3 )]联合阿糖胞苷20 mg/m 2皮下注射每天一次在每28天的第1-10天- 天周期从第 1 周期第 1 天开始。在加速期间,患者接受 TLS 预防并住院进行监测。一旦骨髓评估确认缓解(定义为第 1 周期治疗后白血病原始细胞少于 5% 并伴有血细胞减少),VENCLEXTA 中断长达 14 天,或直至 ANC ≥500/微升且血小板计数≥50 × 10 3/微升。患者继续治疗直至疾病进展或出现不可接受的毒性。基线人口统计和疾病特征如表29所示。
表 29. 接受 VENCLEXTA 联合低剂量阿糖胞苷治疗的 AML 患者的基线患者特征
中位随访时间为 7.3 个月(范围:0.3 至 54.0 个月)。CR 率为 21% (95% CI: 12, 34),CRh 率为 21% (95% CI: 12, 34)。
CR 的中位持续时间为 22.9 个月(95% CI:5.1,-),CR+CRh 的中位持续时间为 14.3 个月(95% CI:6.1,31.2)。
使用 VEN+LDAC 治疗的患者达到首次 CR 或 CRh 的中位时间为 1.0 个月(范围:0.8 至 9.4 个月)。
该试验另外招募了 21 名患者(年龄范围:67 至 74 岁),他们没有已知的合并症而无法使用强化诱导化疗,并接受了 VEN+LDAC 治疗。CR 率为 33%(95% CI:15%、57%)。CRh 率为 24%(95% CI:8.2%、47%)。一名患者(4.8%)随后接受了干细胞移植。
16 如何供应/储存和处理
VENCLEXTA 的分配如下:
VENCLEXTA 10 mg薄膜衣片为圆形、双凸形、浅黄色,一侧凹陷有“V”,另一侧凹陷有“10”。
VENCLEXTA 50 mg 薄膜包衣片为椭圆形、双凸形、米色凹陷,一侧有“V”,另一侧有“50”。
VENCLEXTA 100 mg 薄膜包衣片为椭圆形、双凸形、浅黄色,一侧凹陷有“V”,另一侧凹陷有“100”。
在 30°C (86°F) 或以下的温度下存放在原容器中。使用原始容器将其分发给患者以防止受潮。
17 患者咨询信息
建议患者阅读 FDA 批准的患者标签(用药指南)。
肿瘤溶解综合征
告知患者 TLS 的潜在风险,特别是在治疗开始时、加速阶段以及中断后恢复时,并立即报告与此事件相关的任何体征和症状(发烧、发冷、恶心、呕吐、精神错乱、呼吸短促、癫痫发作、心律不齐、尿液颜色深或浑浊、异常疲倦、肌肉疼痛和/或关节不适),请其医疗保健提供者(HCP)进行评估[见警告和注意事项(5.1 ) ]。
建议患者在服用 VENCLEXTA 时每天补充充足的水分,以降低 TLS 的风险。建议每天喝 6 至 8 杯水(总计约 56 盎司)。患者应从首次剂量前2天和当天开始喝水,以及每次增加剂量时[见剂量和给药方法( 2.4 )]。
告知患者预约血液检查或其他实验室测试的重要性[见剂量和给药方法( 2.4 )]。
建议患者可能有必要在医院或医疗办公室服用 VENCLEXTA 以监测 TLS。
中性粒细胞减少症
建议患者如果出现发烧或任何感染迹象,请立即联系其医护人员。告知患者需要定期监测血细胞计数[见警告和注意事项( 5.2 )]。
感染
建议患者如果出现发烧或任何感染迹象,立即联系他们的 HCP [见警告和注意事项 ( 5.3 )]。
药物相互作用
建议患者在 VENCLEXTA 治疗期间避免食用葡萄柚产品、塞维利亚橙子或杨桃。告知患者 VENCLEXTA 可能与某些药物发生相互作用;因此,建议患者告知其医疗保健提供者使用任何处方药、非处方药、维生素和草药产品[见禁忌症( 4 )和药物相互作用( 7.1 )]。
免疫接种
忠告患者避免用活疫苗接种,因为用VENCLEXTA治疗期间它们可能不安全或有效[见警告和注意事项( 5.4 )]。
胚胎-胎儿毒性
告知孕妇对胎儿的潜在风险。建议女性或有生育潜力的女性告知其医疗保健提供者已知或怀疑怀孕的情况[参见警告和注意事项( 5.5 )和在特定人群中的使用( 8.1 )]。
建议有生育潜力的女性患者在治疗期间和最后一次剂量后30天内使用有效的避孕措施[见特殊人群中使用( 8.3 )]。
哺乳期
建议妇女用VENCLEXTA治疗期间和末次剂量后1周内不要母乳喂养[见特殊人群中使用( 8.2 )]。
不孕症
忠告生殖潜力男性VENCLEXTA可能损害生育力[见特殊人群中使用( 8.3 )]。
行政
建议患者将 VENCLEXTA 保存在原始容器中。
建议患者完全按照处方服用 VENCLEXTA,不要改变剂量或停止服用 VENCLEXTA,除非他们的 HCP 告知他们这样做。建议患者根据其 HCP 的指示,每天约在每天同一时间口服 VENCLEXTA 一次,并且药片应随餐和水整个吞服,不得咀嚼、压碎或折断[见剂量和给药方法(2.8 )]。
建议 CLL/SLL 患者在治疗的前 4 周内将 VENCLEXTA 保留在原包装中,并且不要将药片转移到不同的容器中。
建议患者如果错过一剂 VENCLEXTA 的时间少于 8 小时,请立即服用错过的剂量,并照常服用下一剂。如果VENCLEXTA错过一剂超过8小时,建议患者等待并在通常时间服用下一剂[见剂量和给药方法( 2.8 )]。
建议患者如果服用 VENCLEXTA 后呕吐,当天不要服用任何额外剂量,并在第二天的通常时间服用下一剂。
制造和销售:
AbbVie Inc.
North Chicago, IL 60064
和
营销商:
Genentech USA, Inc.
罗氏集团成员
South San Francisco, CA 94080-4990
© 2016-2022 AbbVie Inc.
© 2016-2022 Genentech, Inc.
20070720 R1

NDC 0074-0579-28
CLL/SLL 起始包
文克莱斯塔®
(维奈托克片)
10 毫克、50 毫克和 100 毫克
起始包
!警告
当您收到这种药物时,请联系您的医生。
可能需要在医生在场的情况下服用第一剂,以防止潜在的严重副作用。
分配器:每次分配 VENCLEXTA 时,请向患者提供随附的用药指南。
艾伯维
仅接收
基因泰克公司
NDC 0074-0561-14
仅接收
文克莱斯塔®
(维奈托克片)
每片10 毫克
14 片
向每位患者分发随附的用药指南。
艾伯维
基因泰克公司

NDC 0074-0566-11
仅接收
文克莱斯塔®
(维奈托克片)
每片50 毫克
1 片
向每位患者分发随附的用药指南。
艾伯维
基因泰克公司
NDC 0074-0576-22
仅接收
文克莱斯塔®
(维奈托克片)
100毫克
注意:将 Venclexta 分配并储存在原始容器中以防止受潮。
120 片
向每位患者分发随附的用药指南。
艾伯维
基因泰克

NDC 0074-0576-11
仅接收
文克莱斯塔®
(维奈托克片)
每片100 毫克
1 片
向每位患者分发随附的用药指南。
艾伯维
基因泰克

NDC 0074-0576-30®
仅接收
文克莱斯塔
(维奈托克片)
100毫克
注意:将 Venclexta 分配并储存在原始容器中以防止受潮。
28 片
向每位患者分发随附的用药指南。
艾伯维
基因泰克公司

【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
本说明书来源于:美国FDA
https://nctr-crs.fda.gov/fdalabel/services/spl/set-ids/b118a40d-6b56-cee3-10f6-ded821a97018/spl-doc?hl=venetoclax
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书



NDC 0074-0579-28
CLL/SLL Starting Pack
VENCLEXTA®
(venetoclax tablets)
10 mg, 50 mg, and 100 mg
Starting Pack
! WARNING
Contact your doctor when you receive this medication.
It may be necessary to take your first dose in the presence of your doctor to prevent a potential serious side effect.
DISPENSER: Each time VENCLEXTA is dispensed give the patient the enclosed Medication Guide.
abbvie
Rx only
Genentech
NDC 0074-0561-14
Rx only
VENCLEXTA®
(venetoclax tablets)
10 mg per tablet
14 Tablets
Dispense the accompanying Medication Guide to each patient.
abbvie
Genentech

NDC 0074-0566-11
Rx only
VENCLEXTA®
(venetoclax tablets)
50 mg per tablet
1 Tablet
Dispense the accompanying Medication Guide to each patient.
abbvie
Genentech
NDC 0074-0576-22
Rx only
VENCLEXTA®
(venetoclax tablets)
100 mg
Attention: Dispense and store Venclexta in original container to protect from moisture.
120 Tablets
Dispense the accompanying Medication Guide to each patient.
abbvie
Genetech

NDC 0074-0576-11
Rx only
VENCLEXTA®
(venetoclax tablets)
100 mg per tablet
1 Tablet
Dispense the accompanying Medication Guide to each patient.
abbvie
Genetech
NDC 0074-0576-30®
Rx only
VENCLEXTA
(venetoclax tablets)
100 mg
Attention: Dispense and store Venclexta in original container to protect from moisture.
28 Tablets
Dispense the accompanying Medication Guide to each patient.
abbvie
Genentech

【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。