商品详情
返回产品目录



商品包装及说明书因厂家更换频繁,如有不符以实物为主
维奈克拉片
国际零售参考价:¥**/盒
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。
完整的处方信息
1适应症和用途
1.1慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
VENCLEXTA适用于治疗慢性淋巴细胞性白血病(CLL)或小淋巴细胞性淋巴瘤(SLL)的成年患者。
1.2急性髓细胞白血病
VENCLEXTA与阿扎胞苷,地西他滨或低剂量阿糖胞苷合用,可用于治疗75岁以上或有合并症而不能使用强化诱导化疗的成人的新诊断的急性髓细胞性白血病(AML) 。
该适应症是根据缓解率在加速批准下批准的[请参阅临床研究(14.2)]。对于该适应症的持续批准可能取决于验证试验中对临床益处的验证和描述。
2用法用量
2.1推荐剂量
评估患者特定因素的肿瘤溶解综合征(TLS)风险水平,并在首次服用VENCLEXTA之前为患者提供预防性水合作用和抗高尿酸药物治疗,以降低TLS的风险[参见剂量和给药方法(2.2)和警告和注意事项(5.1)]。
指示患者每天大约在同一时间服用VENCLEXTA片剂,并同时进餐和饮水。VENCLEXTA片剂应完整吞咽,吞咽前不可咀嚼,压碎或弄碎。
慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
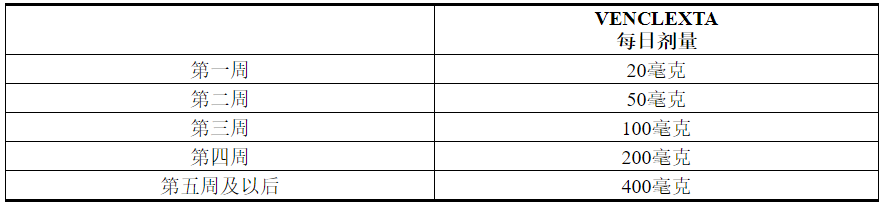
VENCLEXTA的剂量从5周开始增加。
VENCLEXTA 5周剂量递增时间表
如表1所示,按照5周内每周增加的时间表,服用VENCLEXTA剂量至建议的400 mg每日剂量。设计为期5周的加药时间表旨在逐步减少肿瘤负担(散装)并降低TLS的风险。
表1. CLL / SLL患者斜升阶段的给药时间表
根据启动时间表,CLL / SLL入门包提供VENCLEXTA的前4周。使用装在瓶子中的100毫克片剂可以达到400毫克的剂量[请参阅如何提供/储存和处理(16)]。
VENCLEXTA与Obinutuzumab组合
在第1个周期的第1天开始以100 mg的obinutuzumab给药,然后在第1个周期的第2天以900 mg的剂量开始给药。在第1个周期的第8天和第15天以及随后的每个28天周期的第1天,总共给药6 mg周期。请参阅obinutuzumab处方信息以获取推荐的obinutuzumab剂量信息。
在第1天的第22天,按照5周的启动时间表启动VENCLEXTA(请参见表1)。在第2阶段第28天完成增加时间表后,患者应从第3阶段第1天到第12阶段的最后一天每天继续服用VENCLEXTA 400 mg。
VENCLEXTA与利妥昔单抗联合
在患者完成VENCLEXTA的5周剂量递增计划后(见表1),并已接受400 mg VENCLEXTA剂量达7天后,开始利妥昔单抗给药。在每个28天周期的第1天给予利妥昔单抗治疗6个周期,其中利妥昔单抗的第1周期静脉给药剂量为375 mg / m 2,第2-6 周期的静脉内给药剂量为500 mg / m 2。
自利妥昔单抗的第1周期第1天起,患者应连续24个月每天持续服用VENCLEXTA 400 mg,持续24个月。
VENCLEXTA作为单药治疗
VENCLEXTA的推荐剂量是在患者完成5周剂量递增计划后每天一次。VENCLEXTA应该每天口服一次,直到观察到疾病进展或出现不可接受的毒性。
急性髓性白血病
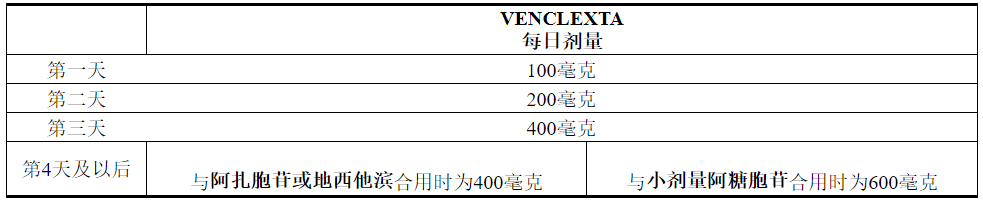
VENCLEXTA的剂量取决于组合剂。
VENCLEXTA的投药时间表(包括加药量)如表2所示。在第1天开始使用阿扎胞苷或地西他滨或小剂量阿糖胞苷。
表2. AML患者准备阶段的给药时间表
继续VENCLEXTA,与阿扎胞苷或地西他滨或小剂量阿糖胞苷合用,直至观察到疾病进展或不可接受的毒性。
2.2肿瘤溶解综合征的风险评估和预防
用VENCLEXTA治疗的患者可能会发生肿瘤溶解综合征。有关管理的特定详细信息,请参阅下面的相应部分。评估患者特定因素的肿瘤溶解综合征(TLS)风险水平,并在首次服用VENCLEXTA之前为患者提供预防性水合作用和抗高尿酸药,以降低TLS风险。
慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
VENCLEXTA可以导致肿瘤迅速减少,因此在开始的5周加速阶段中会产生TLS风险。与TLS相一致的血液化学变化需要及时管理,最早可在VENCLEXTA首次给药后6到8个小时内发生,并且每次剂量增加时都会发生。
TLS的风险是基于多种因素的连续性,包括肿瘤负担和合并症。肾功能降低(肌酐清除率[CLcr] <80 mL / min)进一步增加了风险。在开始使用VENCLEXTA治疗之前,对所有患者进行肿瘤负荷评估,包括放射线评估(例如CT扫描),评估血液化学(钾,尿酸,磷,钙和肌酐),并纠正先前存在的异常。随着肿瘤负担的减少,风险可能降低[见警告和注意事项(5.1)和在特定人群中的使用(8.6)]。
下表3描述了根据临床试验数据确定的肿瘤负荷,在VENCLEXTA治疗期间建议的TLS预防和监测。在最终确定预防措施和监测时间表之前,请考虑所有患者合并症。
表3.基于CLL / SLL患者的肿瘤负担的建议TLS预防
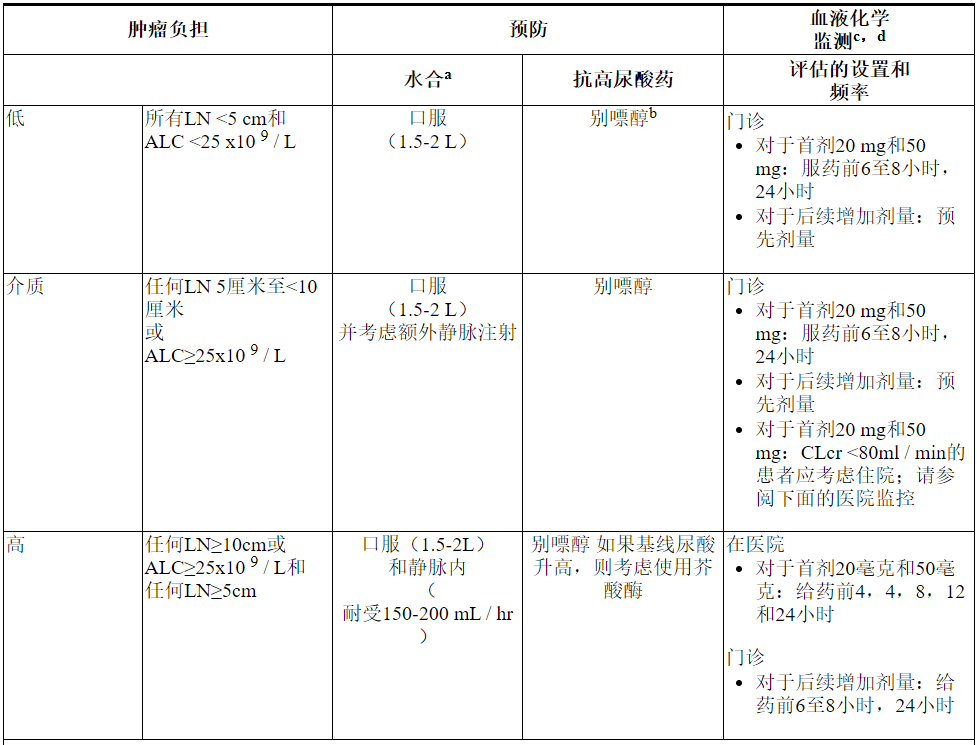

急性髓性白血病
- 在开始VENCLEXTA之前,所有患者的白细胞计数应小于25×10 9 / L。治疗前可能需要进行细胞还原。
- 在首次服用VENCLEXTA之前,为所有患者提供预防措施,包括适当的水合作用和抗高尿酸药,并在逐步增加阶段继续使用。
- 在开始使用VENCLEXTA治疗之前,评估血液化学成分(钾,尿酸,磷,钙和肌酐)并纠正先前存在的异常。
- 在服药前,每次新药剂量增加后6至8小时以及达到最终剂量后24小时,监测血液化学成分中的TLS。
- 对于存在TLS危险因素的患者(例如循环母细胞,白血病累及骨髓的高负担,治疗前乳酸脱氢酶(LDH)水平升高或肾功能下降),应考虑采取其他措施,包括增加实验室监测和减少VENCLEXTA的启动剂量。
2.3基于毒性的剂量修改
慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
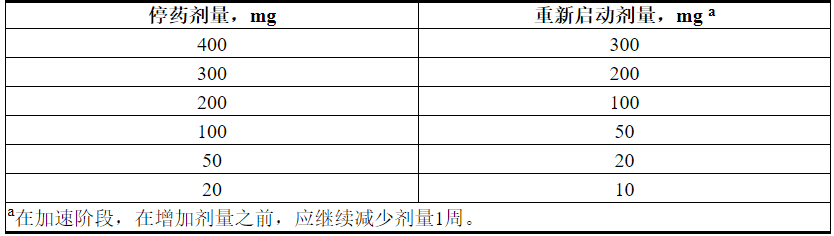
中断给药或减少毒性剂量。有关与VENCLEXTA相关的毒性的推荐剂量修改,请参见表4和表5。对于在启动阶段的前5周内给药中断大于1周或在完成启动阶段后大于2周的给药中断患者,应重新评估TLS的风险,以确定是否需要降低剂量重新开始(例如,所有或剂量斜坡上升时间表的一些水平)[见剂量和给药(2.1,2.2)]。
表4.针对CLL / SLL中毒性a的推荐VENCLEXTA剂量修改
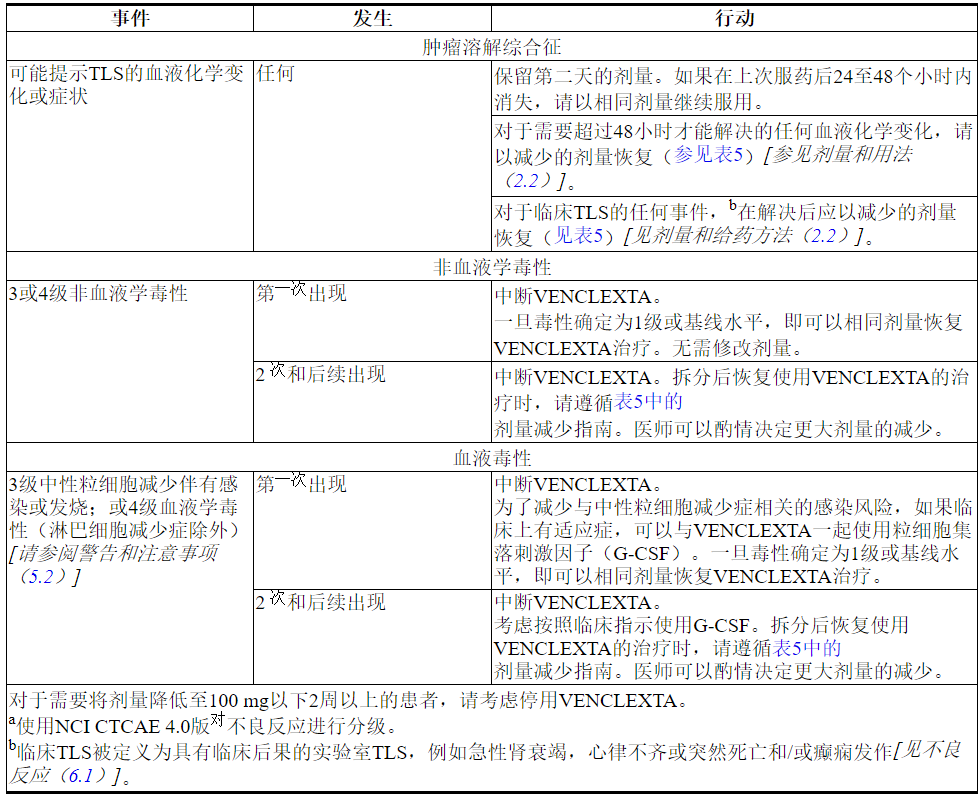
表5.在CLL / SLL中进行VENCLEXTA治疗期间降低的毒性剂量
急性髓性白血病
经常通过减少血细胞减少症来监测血球计数。处理某些不良反应[请参阅警告和注意事项(5.2)和不良反应(6.2)]可能需要中断剂量或永久停用VENCLEXTA。表6显示了血液毒性的剂量调整指南。
表6. AML中毒性a的推荐剂量修改
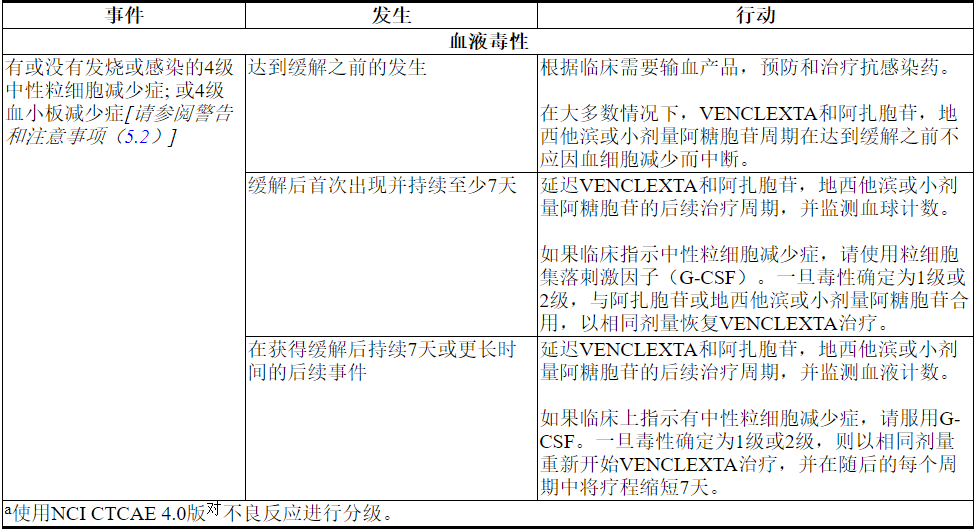
2.4与强或中度CYP3A抑制剂或P-gp抑制剂同时使用的剂量修改
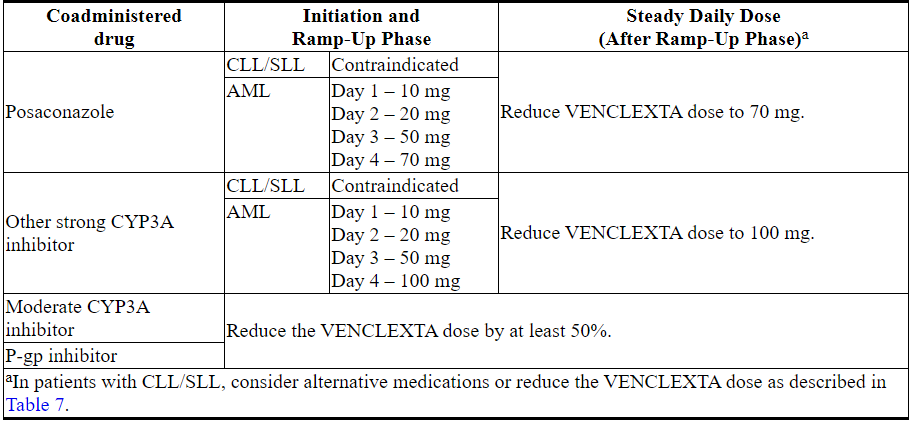
表7描述了在启动阶段,启动阶段或启动阶段期间与强或中度CYP3A抑制剂或P-gp抑制剂[见药物相互作用(7.1)]并用的VENCLEXTA禁忌症或剂量调整。
在抑制剂停止后2至3天恢复在强效或中度CYP3A抑制剂或P-gp抑制剂同时使用之前的VENCLEXTA剂量[见剂量和给药方法(2.3)和药物相互作用(7.1)]。
表7.潜在的VENCLEXTA与CYP3A和P-gp抑制剂的相互作用的管理
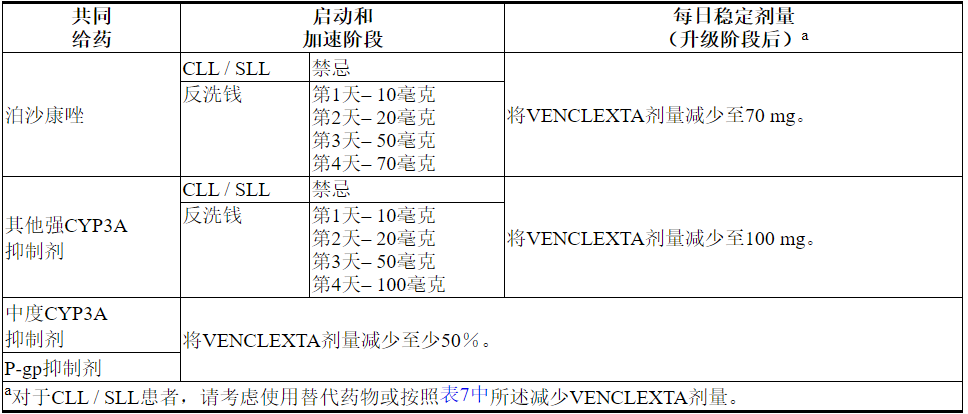
2.5重度肝功能不全患者的剂量调整
对于严重肝功能不全的患者,将VENCLEXTA每日一次剂量降低50%(Child-Pugh C);更加密切地监测这些患者的毒性征兆[请参阅在特定人群中使用(8.7)和临床药理学(12.3)]。
2.6剂量遗漏
如果患者在通常服用8个小时内错过VENCLEXTA剂量,则患者应尽快服用错过的剂量并恢复正常的每日给药计划。如果患者错过了超过8个小时的剂量,则患者不应服用错过的剂量,而应在第二天恢复常规给药时间表。
如果患者在服药后呕吐,则当天不应再服用其他剂量。下一次处方剂量应在通常时间服用。
3剂型和强度
表8. VENCLEXTA片剂的强度和说明

4禁忌症
在CLL / SLL患者中,VENCLEXTA与强 CYP3A抑制剂的同时使用在CLL / SLL患者中是禁忌的,因为它可能增加肿瘤溶解综合征的风险[见剂量和给药方法(2.4)和药物相互作用(7.1)] ]。
5警告和注意事项
5.1肿瘤溶解综合征
肿瘤溶解综合征(TLS),包括致命性事件和肾功能衰竭需要透析,当VENCLEXTA处理发生在患者的肿瘤负荷高[见不良反应(6.1,6.2)]。
在遵循当前(5周)剂量增加以及TLS预防和监测措施的CLL患者中,VENCLEXTA CLL单药研究的TLS发生率为2%。TLS的发生率与VENCLEXTA和obinutuzumab或rituximab的组合保持一致。随着CLL / SLL患者2至3周剂量的增加和更高的起始剂量,TLS发生率为13%,包括死亡和肾衰竭[见不良反应(6.1)]。
VENCLEXTA可以引起肿瘤的快速减少,因此在启动阶段和启动阶段会造成TLS风险。与TLS相一致的血液化学变化需要及时管理,最早可在VENCLEXTA首次给药后6到8个小时内发生,并且每次剂量增加时都会发生。
TLS的风险是基于多种因素的连续性,包括肿瘤负担和合并症。肾功能下降进一步增加了患病风险。应该对患者进行风险评估,并应该对TLS进行适当的预防,包括水合作用和抗高尿酸药。监测血液化学成分并及时处理异常。必要时中断加药。聘请更多的密集措施(静脉补液,频繁的监测,住院)作为整体风险的增加[见剂量和给药方法(2.2,2.3)和特殊人群中使用(8.6)]。
VENCLEXTA与P-gp抑制剂或强或中度CYP3A抑制剂同时使用会增加venetoclax暴露,可能会增加TLS在开始时和启动阶段的风险,并需要VENCLEXTA剂量调整[参见剂量和给药方法(2.4)和药物相互作用(7.1)]。
5.2中性粒细胞减少
在CLL患者,3或4级中性白细胞减少症,63%发展到64%的患者和4级中性白细胞减少在31%发展到与VENCLEXTA组合和单一疗法研究中治疗的患者的33%(见表10,12,14)。联合和单药治疗研究中,接受VENCLEXTA治疗的患者中有4%至6%出现发热性中性粒细胞减少症[见不良反应(6.1)]。
在AML患者中,接受VENCLEXTA与阿扎胞苷或地西他滨或小剂量阿糖胞苷合用的VENCLEXTA治疗的患者中,中性粒细胞计数降低了97%至100%。中性粒细胞减少症可在随后的治疗周期中复发。
在整个治疗期间监控全血细胞计数。严重中性粒细胞减少症的中断给药或减少剂量。考虑采取支持措施,包括用于感染迹象的抗菌剂和使用生长因子(例如G-CSF)[参见剂量和给药方法(2.3)]。
5.3感染
用VENCLEXTA治疗的患者发生了致命的和严重的感染,例如肺炎和败血症[见不良反应(6.1)]。密切监视患者的感染迹象和症状,并及时治疗。对于第3级和更高级别的感染,不使用VENCLEXTA [请参阅剂量和用法(2.3)]。
5.4免疫
在VENCLEXTA治疗之前,之中或之后,不要给予减毒活疫苗,直到B细胞恢复。尚未研究VENCLEXTA治疗期间或之后用减毒活疫苗免疫的安全性和有效性。劝告患者接种疫苗可能无效。
5.5胚胎-胎儿毒性
根据其作用机理和在动物中的发现,VENCLEXTA对孕妇给药可能会引起胚胎胎儿伤害。在对小鼠进行的胚胎-胎儿研究中,以每天400 mg的剂量与孕妇体内观察到的暴露量相等的静脉内注射给怀孕的动物,导致植入后损失和胎儿体重下降。在使用VENCLEXTA的孕妇中,尚无充分且对照良好的研究。建议有生殖能力的女性在治疗期间避免怀孕。如果在怀孕期间使用VENCLEXTA或患者在服用VENCLEXTA期间怀孕,则应告知患者对胎儿的潜在危害[请参见在特定人群中使用(8.1) ]。
5.6当在硼替佐米和地塞米松中添加VENCLEXTA时,多发性骨髓瘤患者的死亡率增加
在患有复发性或难治性多发性骨髓瘤的患者的一项随机试验(BELLINI; NCT02755597)中,在硼替佐米加地塞米松中添加VENCLEXTA(未表明使用VENCLEXTA会导致死亡率增加)。在对照临床试验之外,不建议将VENCLEXTA与硼替佐米联合地塞米松联合治疗多发性骨髓瘤。
6不良反应
标签的其他部分详细讨论了以下临床上显着的不良反应:
- 肿瘤溶解综合征[请参阅警告和注意事项(5.1)]
- 中性粒细胞减少症[请参阅警告和注意事项(5.2)]
- 感染[请参阅警告和注意事项(5.3)]
由于临床试验是在变化很大的条件下进行的,因此不能将一种药物的临床试验中观察到的不良事件发生率与另一种药物的临床试验发生率直接进行比较,并且可能无法反映实际中观察到的发生率。
6.1慢性淋巴细胞白血病/小淋巴细胞淋巴瘤的临床试验经验
CLL14
在一项随机,开放标签,主动对照试验中,对以前未经治疗的CLL患者,评估了VENCLEXTA联合obinutuzumab(VEN + G)与obinutuzumab联合苯丁酸氮芥(GClb)的安全性。
随机分配给VEN + G组的患者接受VENCLEXTA和obinutuzumab联合治疗六个周期,然后接受VENCLEXTA作为单药治疗另外六个周期。患者在第1个周期的第22天开始进行VENCLEXTA的5周增加剂量的第一剂,一旦完成,每天继续一次VENCLEXTA 400 mg,共12个周期。研究治疗的详细信息在第14节中描述[请参见临床研究(14.1)]。该试验要求总累积疾病评分量表(CIRS)> 6或CLcr <70 mL / min,肝转氨酶和总胆红素≤正常上限的2倍,并排除CIRS单个器官/系统损伤评分为4的患者除了眼,耳,鼻和喉器官系统。
总共治疗了426名患者(VEN + G组212名,GClb组214名)。VENCLEXTA的中位暴露时间为10.5个月(范围:0到13.5个月)。奥比妥珠单抗的中位循环数为6,苯丁酸氮芥的中位循环数为12。
在VEN + G组中,有2%(4/212)的患者报告了在没有疾病进展的情况下且在上次研究治疗后28天内发作的致命不良反应,大多数是感染引起的。据报道,VEN + G组中有49%的患者出现严重不良反应,最常见的原因是发热性中性粒细胞减少和肺炎(各占5%)。
在VEN + G组中,不良反应导致16%的患者停止治疗,减少21%的剂量和74%的剂量中断。在VEN + G组中,中性粒细胞减少症导致VENCLEXTA的剂量中断在41%的患者中减少了13%,并中断了2%。
表9和表10分别显示了CLL14试验中确定的不良反应和实验室异常。VEN + G最常见(≥15%)的不良反应是中性粒细胞减少,腹泻,疲劳,恶心,贫血和上呼吸道感染。
表9.用VEN + G治疗的患者常见(≥10%)不良反应
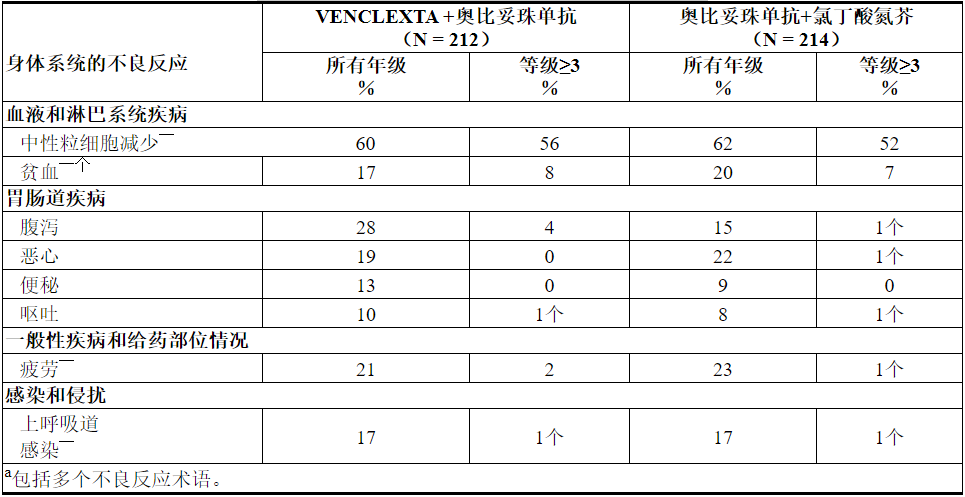
在VEN + G治疗的患者中,<10%报道的其他临床上重要的不良反应(所有级别)如下:
血液和淋巴系统异常:发热性中性粒细胞减少症(6%)
感染和感染(均包括多个不良反应):肺炎(9%),尿路感染(6%),败血症(4%)
代谢与营养失调:肿瘤溶解综合征(1%)
在完成VEN + G联合治疗后,单药VENCLEXTA治疗期间,最常见的全等级不良反应(≥10%患者)为中性粒细胞减少(26%)。最常见的≥3级不良反应(≥2%患者)为中性粒细胞减少症(23%)和贫血(2%)。
表10.用VEN + G治疗的患者中≥10%的新的或重要的临床重要实验室异常发生
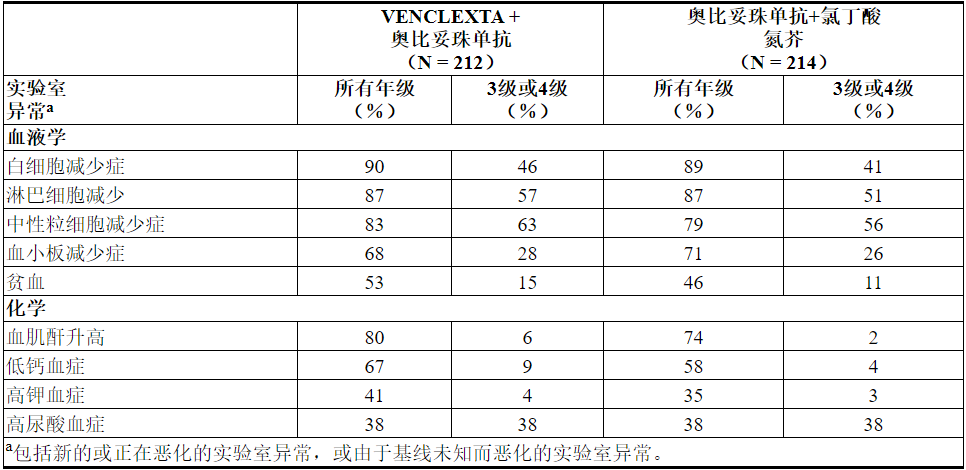
在接受VEN + G治疗的≥2%的患者中发生的4级实验室异常包括中性粒细胞减少症(32%),白细胞减少症和淋巴细胞减少症(10%),血小板减少症(8%),低钙血症(8%),高尿酸血症(7%),血液肌酐增加(3%),高钙血症(3%)和低钾血症(2%)。
村野
在一项开放标签的随机研究中,对接受过至少一种先前治疗的CLL患者评估了VENCLEXTA联合利妥昔单抗(VEN + R)与苯达莫司汀联合利妥昔单抗(B + R)的安全性。
随机分组接受VEN + R的患者完成了计划的加量治疗(5周),并每天一次接受VENCLEXTA 400 mg联合利妥昔单抗的治疗,共6个周期,随后单药VENCLEXTA总共加药24个月。随机分为B + R的患者接受6个周期(每个周期28天),共6个月。研究治疗的详细信息在第14节中描述[请参见临床研究(14.1)]。
在分析时,VEN + R组的中位暴露时间为22个月,而B + R组为6个月。
在VEN + R组中,有2%(4/194)的患者报告了在没有疾病进展的情况下以及在最后一次VENCLEXTA治疗后30天内和/或最后一次利妥昔单抗90天之内发生的致命不良反应。据报道,VEN + R组中有46%的患者出现严重不良反应,其中最常见(≥5%)的是肺炎(9%)。
在VEN + R组中,不良反应导致16%的患者停止治疗,减少15%的剂量和71%的剂量中断。在B + R组中,不良反应导致10%的患者停止治疗,减少15%的剂量和40%的剂量中断。在VEN + R组中,中性粒细胞减少症导致46%的患者中断VENCLEXTA的剂量,并导致3%的患者中断,而血小板减少症导致3%的患者中断。
表11和表12分别显示了MURANO试验中确定的不良反应和实验室异常。MURANO试验并非旨在证明VEN + R的不良反应率与B + R相比,在任何特定的不良反应或实验室异常方面均具有统计学上的显着差异。
表11.与B + R相比,VEN + R治疗的患者报告的常见(≥10%)不良反应为全等级≥5%或≥2%≥3更高发生率
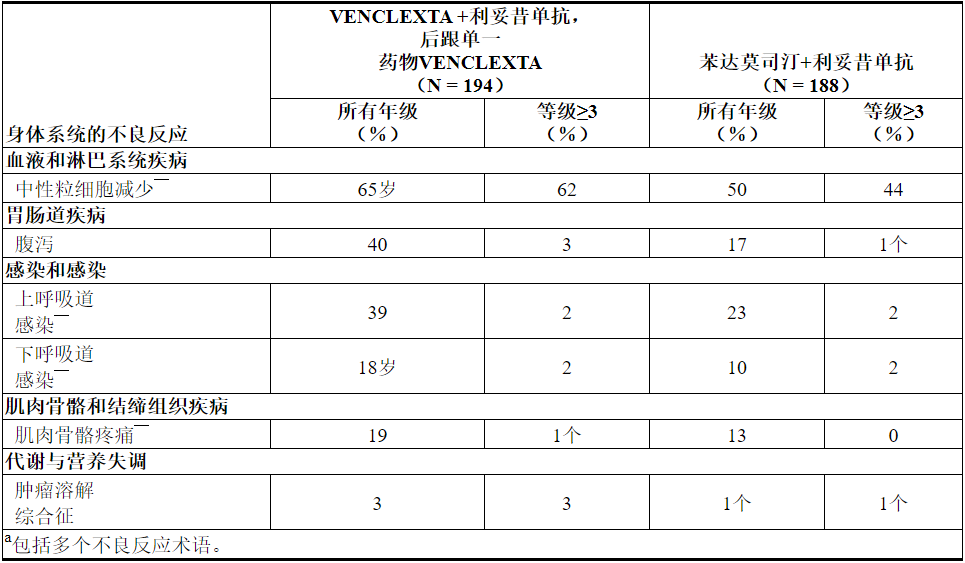
在MURANO的VEN + R组中,≥10%的患者报告了其他不良反应(所有级别),以下是其他重要的不良反应:
血液和淋巴系统疾病:贫血(16%),血小板减少症(15%),发热性中性粒细胞减少症(4%)
胃肠道疾病:恶心(21%),便秘(14%),腹痛(13%),粘膜炎(10%),呕吐(8%)
呼吸系统疾病:咳嗽(22%)
一般疾病和给药部位情况:疲劳(22%),发热(15%)
皮肤疾病:皮疹(13%)
神经系统和精神疾病:头痛(11%),失眠(11%)
感染和侵染:肺炎(10%),败血症(1%)
在完成VEN + R联合治疗后,单药VENCLEXTA治疗期间,报告的最常见的全等级不良反应(≥10%患者)为上呼吸道感染(21%),腹泻(19%),中性粒细胞减少(16%)。和下呼吸道感染(11%)。最常见的3级或4级不良反应(≥2%患者)为中性粒细胞减少症(12%)和贫血(3%)。
实验室异常
表12描述了在MURANO试验中发现的常见治疗紧急实验室异常。
表12.与B + R相比,VEN + R发生率更高(≥10%)(≥5%)(任何年级)或≥2%(3或4年级)的新的或恶化的实验室异常
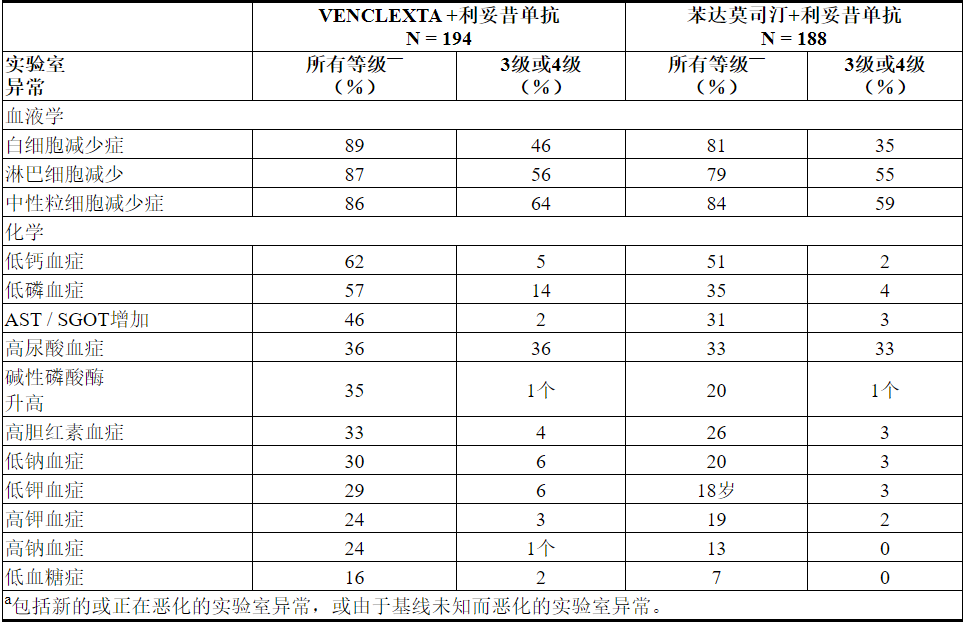
在VEN + R治疗的≥2%的患者中报告的新的4级实验室异常包括中性粒细胞减少症(31%),淋巴细胞减少症(16%),白细胞减少症(6%),血小板减少症(6%),高尿酸血症(4%),低钙血症( 2%),低血糖症(2%)和高镁血症(2%)。
单一疗法研究(M13-982,M14-032和M12-175)
遵循剂量递增时间表后,建议的每日400 mg剂量的单药VENCLEXTA的安全性基于三项单臂试验(M13-982,M14-032和M12-175)的汇总数据。在汇总的数据集中,由352位先前接受过CLL或SLL治疗的患者组成,中位年龄为66岁(范围:28至85岁),白人为93%,男性为68%。先前治疗的中位数为3(范围:0至15)。数据分析时,VENCLEXTA治疗的中位时间为14.5个月(范围:0至50个月)。52%的患者接受VENCLEXTA超过60周。
在VENCLEXTA单药研究中,有2%的患者报告了在无疾病进展且未接受Venetoclax治疗的30天内发生的致命不良反应,最常见的是败血症性休克(2例患者)。据报告,有52%的患者出现严重的不良反应,其中最常见的(≥5%)是肺炎(9%),发热性中性粒细胞减少(5%)和败血症(5%)。
不良反应导致9%的患者停止治疗,降低剂量的13%和中断剂量的36%。导致药物停药的最常见不良反应是血小板减少症和自身免疫性溶血性贫血。导致剂量减少或中断的最常见不良反应(≥5%)是中性粒细胞减少(8%)。
表13列出了在单药VENCLEXTA的这些试验中确定的不良反应。
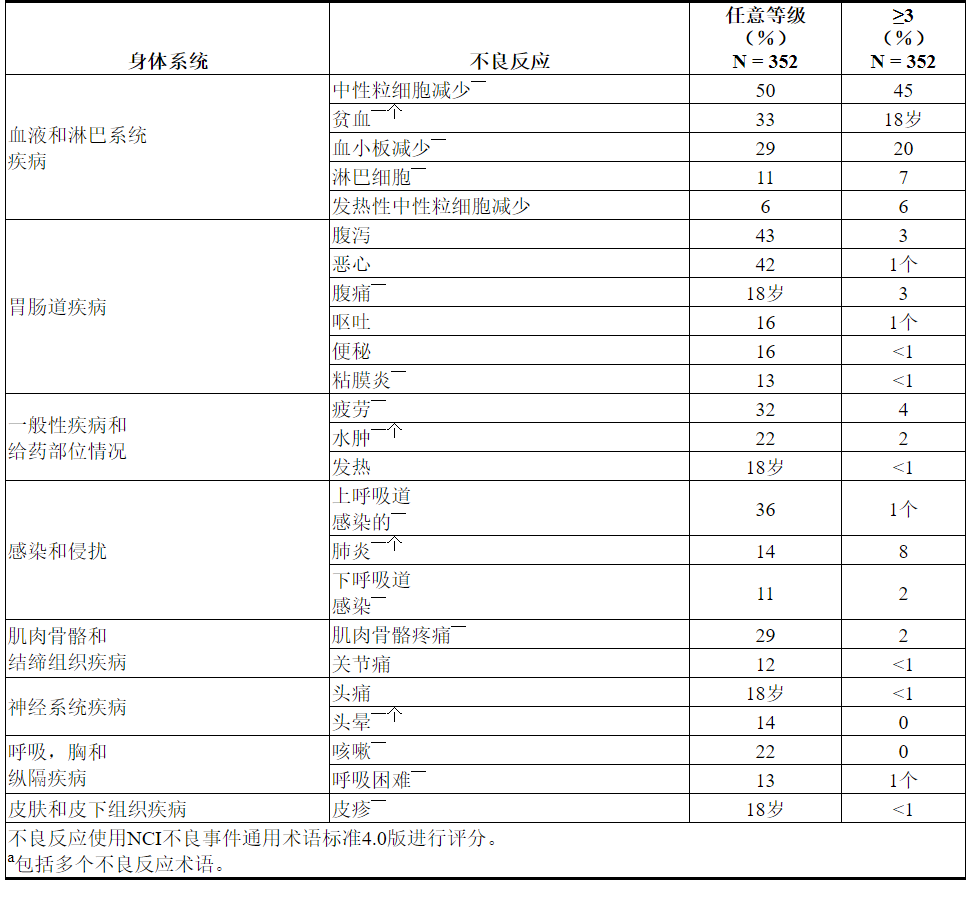
表13.先前治疗过的CLL / SLL(VENCLEXTA单一疗法)的患者中≥10%(任何年级)或≥5%(≥3年级)的不良反应报道
实验室异常
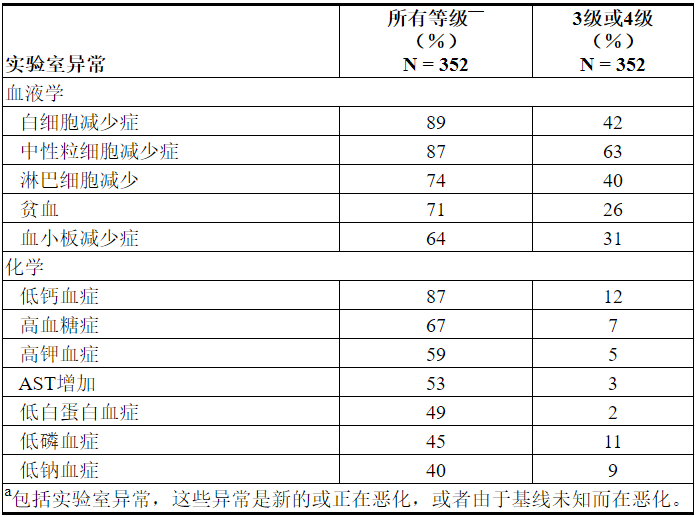
表14描述了整个治疗过程中报告的常见实验室异常,这些异常是新的或比基线恶化的。VENCLEXTA单药治疗最常见的(> 5%)4级实验室异常是血液学实验室异常,包括中性粒细胞减少症(33%),白细胞减少症(11%),血小板减少症(15%)和淋巴细胞减少症(9%)。
表14. VENCLEXTA单药疗法(≥40%任意等级或≥10%的3级或4级)导致的实验室异常或恶化
重要不良反应
肿瘤溶解综合征
启动VENCLEXTA时,肿瘤溶解综合征是确定的重要风险。
CLL14
在接受VEN + G治疗的患者中,TLS的发生率为1%(3/212)[ 参见警告和注意事项(5.1) ]。TLS的所有三个事件均已解决,并且没有导致退出研究。响应TLS事件,奥比努单抗的给药被推迟了两例。
村野
在开放标签的随机第3期研究中,接受VEN + R治疗的患者的TLS发生率为3%(6/194)。后三百八十九分之七十七患者在研究中招募,协议进行了修正以纳入当前TLS预防和监测措施在部分2.1和2.2中描述[见剂量和给药(2.1,2.2)]。TLS的所有事件都发生在VENCLEXTA加速期间,并在两天内得到解决。所有六名患者均完成了加量试验,并达到建议的每日剂量400毫克VENCLEXTA。遵循当前第5周增加时间表和TLS预防和监测措施的患者,在2.1和2.2节中没有观察到临床TLS [请参阅剂量和给药方法(2.1,2.2)]。表12列出了接受VEN + R治疗的患者与TLS相关的实验室异常率。
单一疗法研究(M13-982和M14-032)
在168名CLL患者根据在2.1和2.2节中描述的建议处理的,TLS率为2%[见剂量和给药(2.1,2.2)]。所有事件均符合实验室TLS标准(实验室异常彼此之间在24小时内满足以下≥2的条件:钾> 6 mmol / L,尿酸> 476 µmol / L,钙<1.75 mmol / L或磷> 1.5 mmol / L); 或被报告为TLS事件。该事件发生在淋巴结≥5cm和/或ALC≥25x 10 9的患者中/升 所有事件均在5天内解决。在这些患者中,未观察到具有急性肾衰竭,心律不齐或猝死和/或癫痫发作等临床后果的TLS。所有患者的CLcr≥50mL / min。与TLS相关的实验室异常为高血钾症(所有等级17%,1%≥3级),高磷酸盐血症(所有等级14%,2%≥3级),低钙血症(16%所有等级,2%≥3级)和高尿酸血症(所有等级的10%,<1%的等级≥3)。
在最初的1期剂量寻找试验中,启动阶段较短(2-3周),起始剂量较高,TLS的发生率为13%(10/77; 5个实验室TLS,5个临床TLS),包括2例致命事件和3例急性肾衰竭事件,其中1例需要透析。这方面的经验后,TLS风险评估,给药方案,TLS预防和修订了监测措施[见剂量和给药方法(2.1,2.2)]。
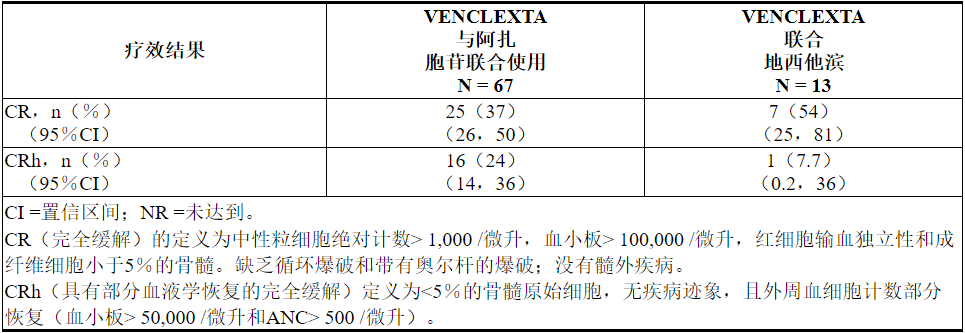
6.2急性髓细胞性白血病的临床试验经验
VENCLEXTA(400 mg每日剂量)与阿扎胞苷(n = 67)或地西他滨(n = 13)和VENCLEXTA(600 mg每日剂量)结合低剂量阿糖胞苷(n = 61)的安全性基于两个新诊断的AML患者的非随机试验[参见临床研究(14.2)]。服用VENCLEXTA结合阿扎胞苷和地西他滨的患者的中位暴露时间分别为6.5个月(范围:0.1至31.9个月)和8.4个月(范围:0.5至22.3个月)。服用VENCLEXTA联合低剂量阿糖胞苷的患者的中位暴露时间为3.9个月(范围:0.2至29.2个月)。
VENCLEXTA与阿扎胞苷或地西他滨合用
阿扎胞苷
任何等级中最常见的不良反应(≥30%)为恶心,腹泻,便秘,中性粒细胞减少,血小板减少,出血,周围水肿,呕吐,疲劳,发热性中性粒细胞减少,皮疹和贫血。
据报道75%的患者出现严重的不良反应。最严重的严重不良反应(≥5%)是高热性中性粒细胞减少,肺炎(不包括真菌),败血症(不包括真菌),呼吸衰竭和多器官功能障碍综合征。
在开始治疗后的30天内,致命的药物不良反应发生率为1.5%。没有反应发生率≥2%。
21%的患者因不良反应而停药。导致药物停用的最常见不良反应(≥2%)是发热性中性粒细胞减少和肺炎(真菌除外)。
61%的患者发生了由于不良反应引起的剂量中断。导致剂量中断(≥5%)的最常见不良反应是中性粒细胞减少,发热性中性粒细胞减少和肺炎(真菌除外)。
由于不良反应导致的剂量减少发生在12%的患者中。导致剂量减少(≥5%)的最常见不良反应是中性粒细胞减少症。
地西他滨
任何级别最常见的不良反应(≥30%)是高热性中性粒细胞减少,便秘,疲劳,血小板减少,腹痛,头晕,出血,恶心,肺炎(不包括真菌),败血症(不包括真菌),咳嗽,腹泻,中性粒细胞减少,背痛,低血压,肌痛,口咽痛,周围水肿,发热和皮疹。
据报道85%的患者出现严重的不良反应。最常见的严重不良反应(≥5%)是高热性中性粒细胞减少,败血症(不包括真菌),肺炎(不包括真菌),腹泻,疲劳,蜂窝织炎和局部感染。
在开始治疗后的30天内发生了一次(8%)致命的菌血症药物不良反应。
38%的患者因不良反应而停药。导致药物停用(≥5%)的最常见不良反应是肺炎(真菌除外)。
62%的患者发生了由于不良反应引起的剂量中断。导致剂量中断(≥5%)的最常见不良反应是发热性中性粒细胞减少,中性粒细胞减少和肺炎(真菌除外)。
15%的患者因不良反应导致剂量减少。导致剂量减少(≥5%)的最常见不良反应是中性粒细胞减少症。
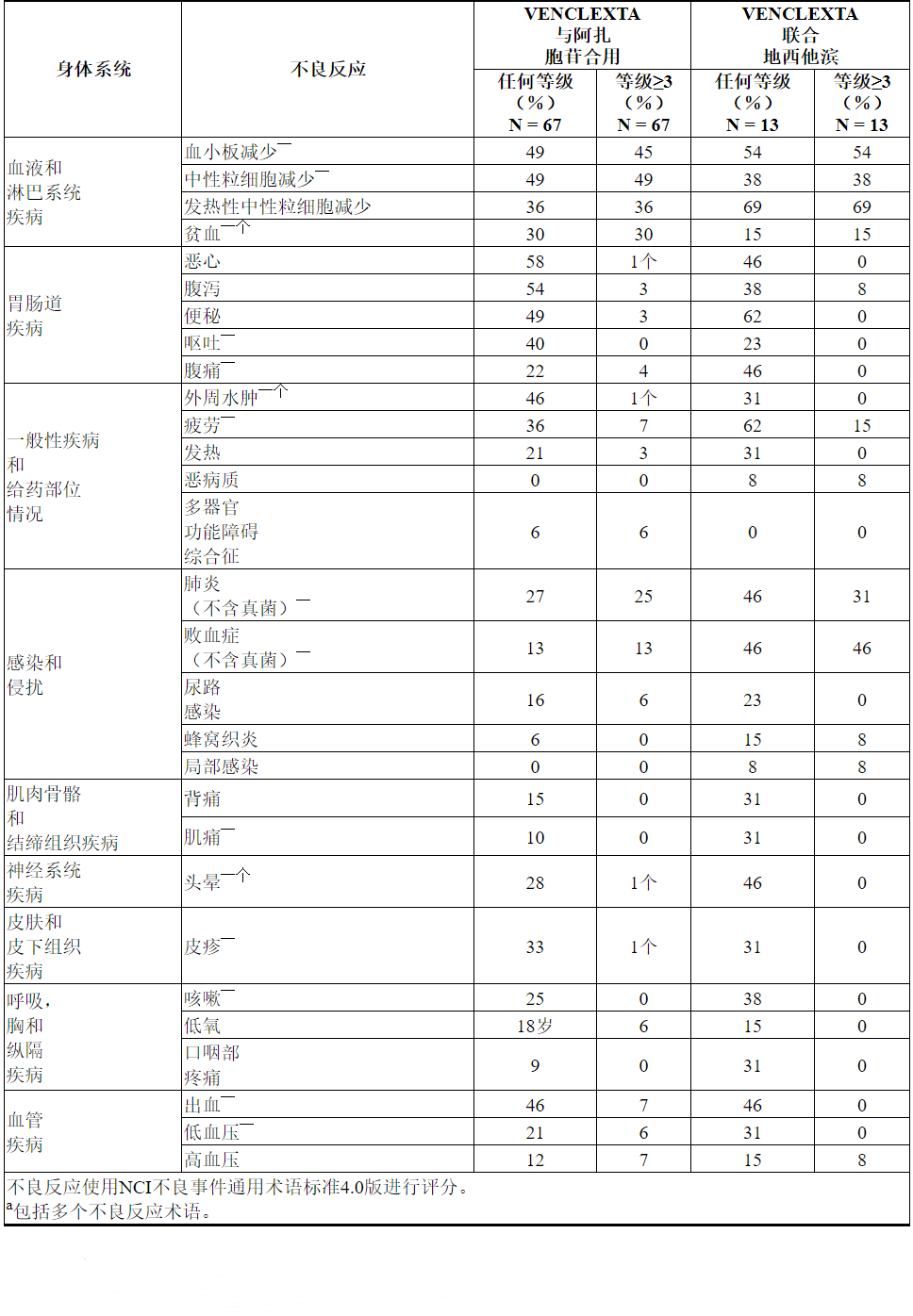
表15列出了新诊断为VENCLEXTA与阿扎胞苷或地西他滨联用的AML患者的不良反应。
表15. VENCLEXTA与阿扎胞苷或地西他滨联合治疗的AML患者中≥30%(任何年级)或≥5%(≥3级)的不良反应报道
实验室异常
表16描述了整个治疗期间报告的常见实验室异常,这些异常是新的或比基线恶化的。
表16. VENCLEXTA联合阿扎胞苷或地西他滨治疗的AML患者中≥40%(任何等级)或≥10%(3或4级)的新出现或恶化的VENCLEXTA实验室异常报道
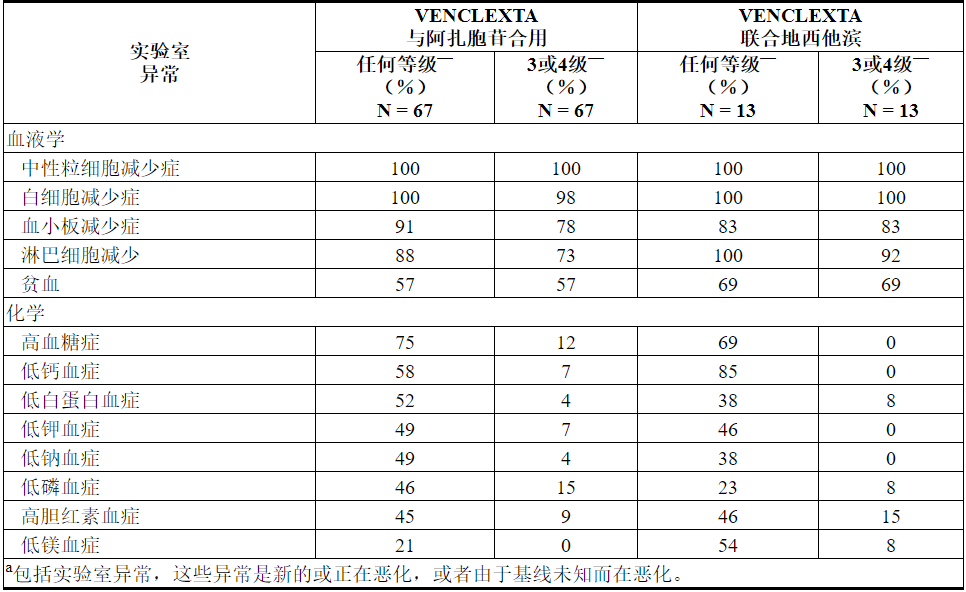
VENCLEXTA与小剂量阿糖胞苷合用
任何等级中最常见的不良反应(≥30%)是恶心,血小板减少,出血,高热性中性粒细胞减少,中性粒细胞减少,腹泻,疲劳,便秘和呼吸困难。
据报道95%的患者出现严重的不良反应。最常见的严重不良反应(≥5%)是高热性中性粒细胞减少,败血症(不包括真菌),出血,肺炎(不包括真菌)和与设备相关的感染。
致命不良药物反应的发生率在开始治疗后的30天内为4.9%,无反应的发生率≥2%。
33%的患者因不良反应而停药。导致药物停用的最常见不良反应(≥2%)是出血和败血症(真菌除外)。
52%的患者因不良反应导致剂量中断。导致剂量中断(≥5%)的最常见不良反应是血小板减少症,中性粒细胞减少和发热性中性粒细胞减少。
8%的患者因不良反应导致剂量减少。导致剂量减少(≥2%)的最常见不良反应是血小板减少症。
表17列出了新诊断为AML并接受VENCLEXTA联合小剂量阿糖胞苷治疗的AML患者的不良反应。
表17. VENCLEXTA联合小剂量阿糖胞苷治疗的AML患者中≥30%(任何年级)或≥5%(≥3级)的不良反应报道
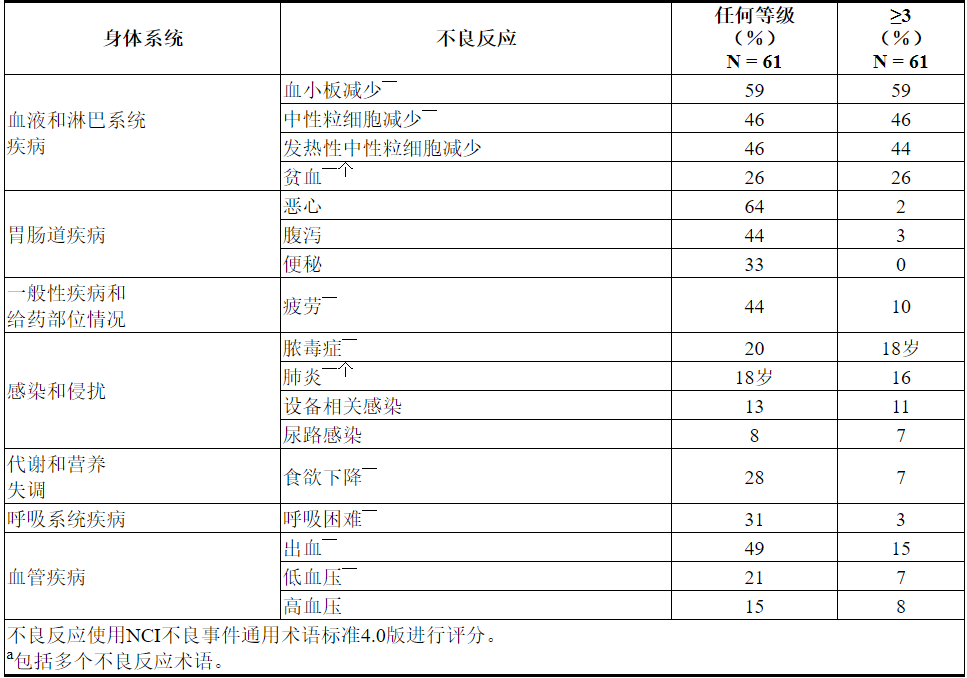
实验室异常
表18描述了整个治疗过程中报告的常见实验室异常,这些异常是新的或从基线开始恶化。
表18. VENCLEXTA联合小剂量阿糖胞苷治疗的AML患者中≥40%(任何级别)或≥10%(3或4级)的新出现或恶化的VENCLEXTA实验室异常报道
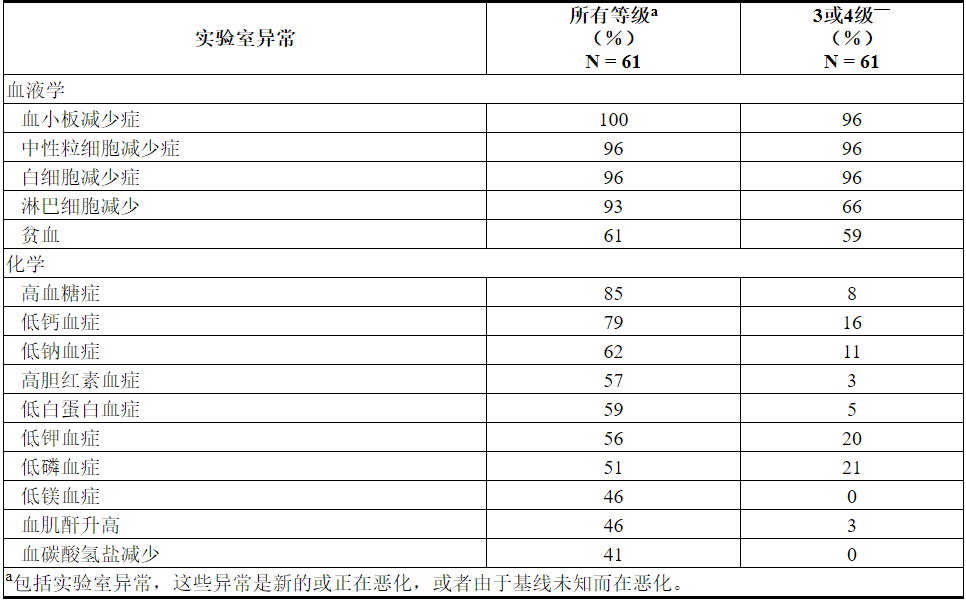
肿瘤溶解综合征
在AML患者中开始治疗时,肿瘤溶解综合征是重要的风险。VENCLEXTA与小剂量阿糖胞苷合用时,TLS发生率为3%(2/61),除了标准的预防和监测措施外,还实施了剂量递增时间表。所有事件均为实验室TLS,所有患者均能够达到目标剂量。
7药物相互作用
7.1其他药物对VENCLEXTA的影响
强或中度CYP3A抑制剂或P-gp抑制剂
与强或中度CYP3A抑制剂或P-gp抑制剂同时使用会增加venetoclax C max和AUC inf [请参阅临床药理学(12.3)],这可能会增加VENCLEXTA毒性,包括TLS的风险[请参阅警告和注意事项(5) ]。
禁忌在CLL / SLL患者开始和上升阶段与强效CYP3A抑制剂同时使用[见禁忌证(4)]。
在患者CLL / SLL服用稳定日剂量(斜坡后阶段),考虑替代药物或调整VENCLEXTA剂量和密切监测VENCLEXTA毒性的体征[见剂量和给药方法(2.3,2.4)]。
在AML患者,调整VENCLEXTA剂量和密切监测VENCLEXTA毒性的体征[见剂量和给药方法(2.3,2.4)]。
恢复已用强或中等CYP3A抑制剂或该抑制剂的停药后一个的P-gp抑制剂2至3天伴随使用之前所用的剂量VENCLEXTA [见剂量和给药(2.3,2.4)]。
在用VENCLEXTA治疗期间,避免使用柚子产品,塞维利亚橙子和杨桃,因为它们含有CYP3A抑制剂。
强或中度CYP3A诱导剂
与强效CYP3A诱导剂同时使用会降低venetoclax C max和AUC inf [参见临床药理学(12.3)],可能会降低VENCLEXTA的疗效。避免将VENCLEXTA与强CYP3A诱导剂或中度CYP3A诱导剂同时使用。
7.2 VENCLEXTA对其他药物的作用
华法林
VENCLEXTA的同时使用会增加华法令C max和AUC inf [参见临床药理学(12.3)],这可能会增加出血的风险。伴有华法林和VENCLEXTA的患者应密切监测国际标准化比率(INR)。
P-gp基板
VENCLEXTA的同时使用会增加P-gp底物的C max和AUC inf [请参阅临床药理学(12.3)],这可能会增加这些底物的毒性。避免将VENCLEXTA与P-gp底物同时使用。如果不可避免地伴随使用,则在VENCLEXTA之前至少6小时分别给P-gp底物配药。
8在特定人群中的使用
8.1怀孕
风险摘要
尚无有关孕妇使用VENCLEXTA的信息,以告知与药物相关的重大先天缺陷和流产风险。根据在小鼠中观察到的毒性,VENCLEXTA对孕妇给药可能会造成胎儿伤害。在小鼠中,venetoclax在每天400 mg人体剂量下的暴露量是人类临床暴露量的1.2倍(基于AUC),具有胎儿毒性。如果在怀孕期间使用VENCLEXTA或患者在服用VENCLEXTA期间怀孕,则应告知患者胎儿的潜在危险。
对于指定人群,估计的主要先天缺陷和流产的背景风险尚不清楚。所有怀孕都有出生缺陷,流产或其他不良后果的背景风险。在美国一般人群中,主要的先天性缺陷的背景风险为临床公认的妊娠的2%至4%,流产为15%至20%。
数据
动物数据
在胚胎胎儿发育研究中,在器官发生期间向怀孕的小鼠和兔子施用了venetoclax。在小鼠中,venetoclax与植入后损失增加和胎儿体重降低(150 mg / kg /天)相关(母体暴露量约为每天400 mg剂量下人 AUC暴露量的1.2倍)。在小鼠或兔子中均未观察到致畸性。
8.2哺乳
风险摘要
没有关于母乳中VENCLEXTA的存在,VENCLEXTA对母乳喂养的孩子的影响或VENCLEXTA对牛奶生产的影响的数据。向哺乳期大鼠给药时,牛奶中存在Venetoclax (参见数据)。
由于许多药物是从人乳中排出的,并且由于VENCLEXTA的母乳喂养的孩子发生严重不良反应的可能性尚不清楚,因此建议哺乳期妇女在VENCLEXTA治疗期间停止母乳喂养。
数据
动物资料
将Venetoclax(单剂量; 150 mg / kg口服)给予产后 8至10天的泌乳大鼠。牛奶中的Venetoclax比血浆中的低1.6倍。母乳中的母体药物(venetoclax)占牛奶中与药物相关的全部物质的大部分,其中三种代谢物含量低。
8.3生殖潜力的男性和女性
VENCLEXTA可能造成胎儿伤害[请参阅警告和注意事项(5.5)和在特定人群中使用(8.1)]。
验孕
在开始服用VENCLEXTA之前,对有生殖潜能的女性进行妊娠试验[见在特定人群中使用(8.1)]。
避孕
劝告有生殖潜力的女性在使用VENCLEXTA治疗期间以及最后一次给药后至少30天使用有效避孕方法 (请参阅“在特定人群中使用(8.1)”)。
不孕症
根据动物的发现,使用VENCLEXTA治疗可能会损害男性的生育能力[参见非临床毒理学(13.1)]。
8.4小儿使用
儿科患者尚未确定安全性和有效性。
在一项青少年毒理学研究中,从7至60天大时通过口服管饲法给小鼠静脉内施用10、30或100 mg / kg /天的venetoclax。毒性的临床体征包括≥30mg / kg /天的活动减少,脱水,皮肤苍白和驼背姿势。此外,死亡率和体重的影响发生在100 mg / kg / day。其他与venetoclax相关的作用是≥10 mg / kg / day的淋巴细胞可逆减少;对于20公斤儿童,以mg / m 2为基础,每天10毫克/千克/天的剂量约为400毫克临床剂量的0.06倍。
8.5老年人使用
慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
在3项VENCLEXTA单项开放性试验的安全性评估中,对352位先前接受过CLL / SLL治疗的患者进行了安全性评估,其中57%(201/352)≥65岁,18%(62/352)≥75岁。
在联合和单药治疗研究中,老年患者和年轻患者之间在安全性和有效性上均未观察到有意义的临床意义差异。
急性髓性白血病
在临床试验中,接受VENCLEXTA联合阿扎胞苷治疗的67例患者中,年龄≥65岁的占96%,年龄≥75岁的占50%。在临床试验中,接受VENCLEXTA联合地西他滨治疗的13例患者中,年龄≥65岁的患者占100%,年龄≥75岁的患者占26%。在接受VENCLEXTA联合小剂量阿糖胞苷治疗的61例患者中,≥65岁的占97%,≥75岁的占66%。
从这些患者中获得了“不良反应和临床研究”部分中提供的功效和安全性数据[参见不良反应(6.2)和临床研究(14.2)]。没有足够的患者人数显示老年患者和年轻患者在安全性和有效性方面的差异。
8.6肾功能不全
由于TLS的风险增加,肾功能降低的患者(CLcr <80 mL / min,由Cockcroft-Gault公式计算)需要更密集的预防和监测,以在开始使用VENCLEXTA治疗时降低TLS的风险[请参阅剂量和用法(2.2,2.3)和警告和注意事项(5.1)]。
建议没有剂量调整为患有轻度或中度肾损伤(CLCR≥30毫升/分钟[见临床药理学(12.3)]。推荐剂量尚未确定为患有严重肾损伤(CLCR <30毫升/分钟)或接受透析的患者。
8.7肝功能不全
对于轻度(Child-Pugh A)或中度(Child-Pugh B)肝功能不全的患者,建议不调整剂量。
减少严重肝功能不全患者的VENCLEXTA剂量(Child-Pugh C);更加密切地监测这些患者的毒性征兆[参见剂量和用法(2.5)和临床药理学(12.3)]。
10过量
VENCLEXTA没有特定的解毒剂。对于服用过量的患者,应密切监测并提供适当的支持治疗;在提升阶段中断VENCLEXTA和监测精心为标志,与其它毒性以及TLS的症状[见剂量和给药方法(2.2,2.3)]。基于Venetoclax的大量分布和广泛的蛋白质结合,透析不太可能导致Venetoclax的大量清除。
11说明
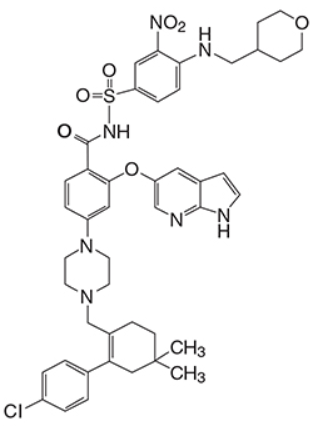
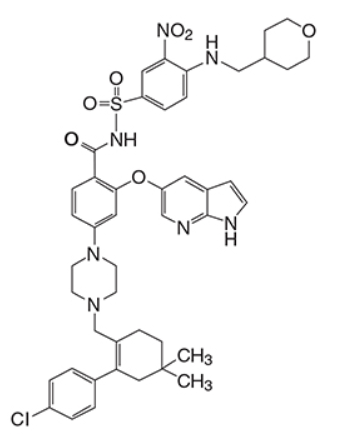
Venetoclax是BCL-2蛋白的选择性抑制剂。它是浅黄色至深黄色固体,其经验式为C 45 H 50 ClN 7 O 7 S,分子量为868.44。Venetoclax具有非常低的水溶性。Venetoclax在化学上被描述为4-(4-{[2-(4-氯苯基)-4,4-二甲基环己-1-烯-1-基]甲基}哌嗪-1-基)-N -({3-硝基-4-[(四氢-2 H-吡喃-4-基甲基)氨基]苯基}磺酰基)-2-(1 H-吡咯并[2,3 - b ]吡啶-5-基氧基)苯甲酰胺)具有以下化学物质结构体:
口服的VENCLEXTA片剂以淡黄色或米色片剂形式提供,其中含有10、50或100 mg 威尼妥克作为活性成分。每片还含有以下非活性成分:共聚维酮,胶体二氧化硅,聚山梨酯80,硬脂富马酸钠和磷酸氢钙。此外,10毫克和100毫克的包衣片剂包括:氧化铁黄,聚乙烯醇,聚乙二醇,滑石粉和二氧化钛。50 mg包衣片剂还包括:氧化铁黄,氧化铁红,氧化铁黑,聚乙烯醇,滑石粉,聚乙二醇和二氧化钛。每个平板的凹坑上刻有“ V”,另一侧刻有“ 10”,“ 50”或“ 100”。
12临床药理学
12.1行动机制
Venetoclax是BCL-2(一种抗凋亡蛋白)的选择性和口服生物利用小分子抑制剂。已经在CLL和AML细胞中证明了BCL-2的过表达,它们在这些细胞中介导肿瘤细胞的存活,并与对化学疗法的抗性有关。Venetoclax通过直接与BCL-2蛋白结合,置换促凋亡蛋白(如BIM),触发线粒体外膜通透性和胱天蛋白酶的活化来帮助恢复凋亡过程。在非临床研究中,venetoclax已证明过表达BCL-2的肿瘤细胞具有细胞毒活性。
12.2药效学
基于疗效的暴露应答分析,在CLL / SLL患者和AML患者的临床研究中观察到药物暴露与更大应答可能性之间的关系。根据安全性的暴露响应分析,在AML患者的临床研究中观察到药物暴露与某些安全事件发生的可能性之间的关系。CLL / SLL患者单药治疗剂量最高至1200 mg,联合利妥昔单抗治疗剂量最高至600 mg,未观察到接触安全性关系。
心脏电生理学
在一项开放标签,单臂研究中,对176例先前接受过血液恶性肿瘤治疗的患者,评估了每天最多一次1200 mg(多于批准的最大推荐剂量的2倍)多剂量VENCLEXTA对QTc间隔的影响。VENCLEXTA对QTc间隔没有大的影响(即> 20 ms),并且静脉胶束暴露与QTc间隔的变化之间没有关系。
12.3药代动力学
Venetoclax稳态平均值(±标准偏差)C max为2.1±1.1 mcg / mL,AUC 0-24为每天服用一次低脂膳食400毫克后为32.8±16.9 mcg•h / mL。Venetoclax稳态AUC在150至800 mg(最大批准推荐剂量的0.25至1.33倍)的剂量范围内成比例增加。Venetoclax的药代动力学不会随时间变化。
吸收性
在喂食条件下多次口服给药后,venetoclax的最大血浆浓度达到5至8小时。
食物的作用
服用低脂餐(约512卡路里,25%脂肪卡路里,60%碳水化合物卡路里和15%蛋白质卡路里)可使Venetoclax暴露量增加约3.4倍,而服用高脂餐(约753卡路里,55 与禁食相比,%脂肪卡路里,28%碳水化合物卡路里和17%蛋白质卡路里)增加了Venetoclax暴露量5.1到5.3倍。
分配
Venetoclax与人血浆蛋白高度结合,血浆中的未结合部分<0.01,浓度范围为1-30微摩尔(0.87-26 mcg / mL)。平均血与血浆之比为0.57。在患者中,venetoclax的表观分布体积(Vd ss / F)为256-321L。
消除
Venetoclax的最终消除半衰期约为26小时。
代谢
Venetoclax 在体外主要被CYP3A代谢。血浆中鉴定出的主要代谢物M27对BCL-2的抑制活性比体外的venetoclax 低至少58倍,其AUC占母体AUC的80%。
排泄
在向健康受试者单次口服放射性标记的[ 14 C] -venetoclax 200 mg后,粪便中回收的剂量> 99.9%(未改变的为20.8%),在9天内尿中的回收率<0.1%。
特定人群
根据年龄(19至90岁),性别,种族(白人,黑人,亚裔和其他),体重,轻度至中度肾功能不全(CLcr 30至89 mL / min),未观察到Venetoclax药代动力学的临床显着差异(由Cockcroft-Gault计算)或轻度至中度肝功能不全(正常总胆红素和天冬氨酸转氨酶(AST)>正常上限(ULN)或总胆红素的1至3倍ULN)。严重肾功能不全(CLcr <30 mL / min)或透析对Venetoclax药代动力学的影响尚不清楚。
肝功能不全患者
服用单剂VENCLEXTA 50 mg后,严重肝功能不全(Child-Pugh C)患者的venetoclax全身暴露(AUC inf)比正常肝功能患者高2.7倍[见剂量和给药方法(2.5)和使用在特定人群中(8.7)]。在轻度或中度肝功能不全的受试者与肝功能正常的受试者之间,未观察到Venetoclax全身暴露的临床相关差异。
药物相互作用研究
临床研究
当与阿扎胞苷,阿奇霉素,阿糖胞苷,地西他滨,胃酸减少剂,奥比妥单抗或利妥昔单抗合用时,未观察到venetoclax药代动力学的临床显着差异。
酮康唑
并用酮康唑(一种强CYP3A,P-gp和BCRP抑制剂)400 mg,每天一次,共7天,可使venetoclax C max升高130%,AUC inf升高540%[参见药物相互作用(7.1)]。
利托那韦
并用ritonavir(一种强效CYP3A,P-gp和OATP1B1 / B3抑制剂)每天一次,持续14天,每天50 mg会使venetoclax C max升高140%,AUC 升高690%[请参见药物相互作用(7.1)]。
泊沙康唑
与单独服用venetoclax 400 mg 相比,将300 mg泊沙康唑(一种强效CYP3A和P-gp抑制剂)与50 mg和100 mg的venetoclax并用7天,分别导致venetoclax C max升高61%和86%。所述venetoclax AUC 24高出90%和144%之间。
利福平
单一剂量利福平的同时使用(一种OATP1B1 / 1B3和P-gp抑制剂)600 mg的Venetoclax C max增加106%,AUC inf增加78%。每天多次服用利福平(作为一种强CYP3A诱导剂)600 mg,连续13天同时使用,会使venetoclax C max降低42%,AUC inf降低71%[见药物相互作用(7.1)]。
华法林
单独使用400 mg 威尼托克与5 mg华法林并用会导致R-华法林和S-华法林的 C max和AUC inf升高18%至28%[请参阅药物相互作用(7.2)]。
地高辛
单独使用100 mg 的Venetoclax与0.5 mg的地高辛(P-gp底物)并用,会使地高辛的C max最高增加35%,AUC inf则增加9% [请参阅药物相互作用(7.2)]。
体外研究
Venetoclax不是CYP1A2,CYP2B6,CYP2C19,CYP2D6或CYP3A4的抑制剂或诱导剂。Venetoclax是CYP2C8,CYP2C9和UGT1A1的弱抑制剂。
Venetoclax不是UGT1A4,UGT1A6,UGT1A9或UGT2B7的抑制剂。
Venetoclax是P-gp和BCRP的抑制剂和底物,是OATP1B1的弱抑制剂。
Venetoclax不是OATP1B3,OCT1,OCT2,OAT1,OAT3,MATE1或MATE2K的抑制剂。
13毒理学
13.1致癌,诱变,生育力受损
venetoclax尚未进行致癌研究。
Venetoclax不是在诱变在体外细菌致突变(Ames的)测定,不诱导数值或结构畸变在一个体外使用人外周血淋巴细胞的染色体畸变试验,并且不是在诱裂体内剂量小鼠骨髓小核试验高达835 mg / kg。在体外 Ames和染色体畸变分析中, M27代谢产物的遗传毒性为阴性。
在雄性和雌性小鼠中进行了生育力和早期胚胎发育研究。这些研究评估了通过植入的交配,受精和胚胎发育。最高剂量为600 mg / kg / day时,venclexlax对发情周期,交配,受精,黄体,子宫植入物或每窝活胚胎没有影响。但是,根据在400 mg剂量下人AUC暴露量低至0.5倍的狗中观察到的睾丸毒性(生殖细胞损失),存在对人类男性生育力的风险。
13.2动物毒理学和/或药理学
在犬中,venetoclax引起包括胆囊,外分泌胰腺和胃在内的各种组织的单细胞坏死,没有证据表明组织完整性或器官功能障碍。这些发现幅度很小至轻微。在4周的给药期和随后的4周恢复期之后,某些组织中仍存在最小的单细胞坏死,并且在较长的给药或恢复期后尚未评估可逆性。
此外,在狗中每天服用约3个月后,由于黑色素的损失,venetoclax导致了毛发的逐渐变白。
14临床研究
14.1慢性淋巴细胞白血病/小淋巴细胞淋巴瘤
联合疗法
CLL14
CLL14(BO25323)是一项随机(1:1),多中心,开放标签,主动控制的3期临床试验(NCT02242942),评估VENCLEXTA联合obinutuzumab(VEN + G)与obinutuzumab联合苯丁酸氮芥的疗效和安全性(GClb)适用于先前未曾接受过CLL治疗且同时患有合并疾病(总累积疾病评分量表[CIRS]评分> 6或CLcr <70 mL / min)的患者。该试验要求肝转氨酶和总胆红素≤正常上限的2倍,并且排除了发生Richter变换或任何CIRS单个器官/系统损伤评分为4的患者(眼,耳,鼻和喉器官系统除外)。
所有患者在第1天(第1天和第2天可分为100 mg和900 mg),第1周期的第8天和第15天以及每个后续周期的第1天接受1000 mg奥比妥珠单抗治疗,共6个周期。在VEN + G组的患者开始5周VENCLEXTA斜坡上升时间表[见剂量和给药(2.1,2.2)]上1个周期的第22天,并得到VENCLEXTA 400 mg,每天一次,从第3个周期第1天,直到最后周期12的第1天。在周期1至12的第1天和第15天,随机分配到GC1b组的患者接受了0.5 mg / kg口服苯丁酸氮芥。
总共432名患者被随机分配,每个研究组216名患者。研究组之间的基线人口统计学和疾病特征相似。中位年龄为72岁(范围:41至89岁),白人占89%,男性占67%;Binet B期和C期分别为36%和43%,东部合作肿瘤小组(ECOG)的表现状态<2为88%。CIRS的中位数为8.0(范围:0至28),58%的患者的CLcr <70 mL / min。在8%的患者中检测到17p缺失,在7%的患者中检测到TP53突变,在19%的患者中检测到11q缺失,在57%的患者中检测到未突变的IgVH。
独立审查委员会(IRC)评估的主要疗效结果为无进展生存期(PFS)。PFS的中位随访时间为28个月(范围:0.1到36个月)。
表19中显示了CLL14的功效结果。PFS的Kaplan-Meier曲线如图1所示。
表19. CLL14的功效结果
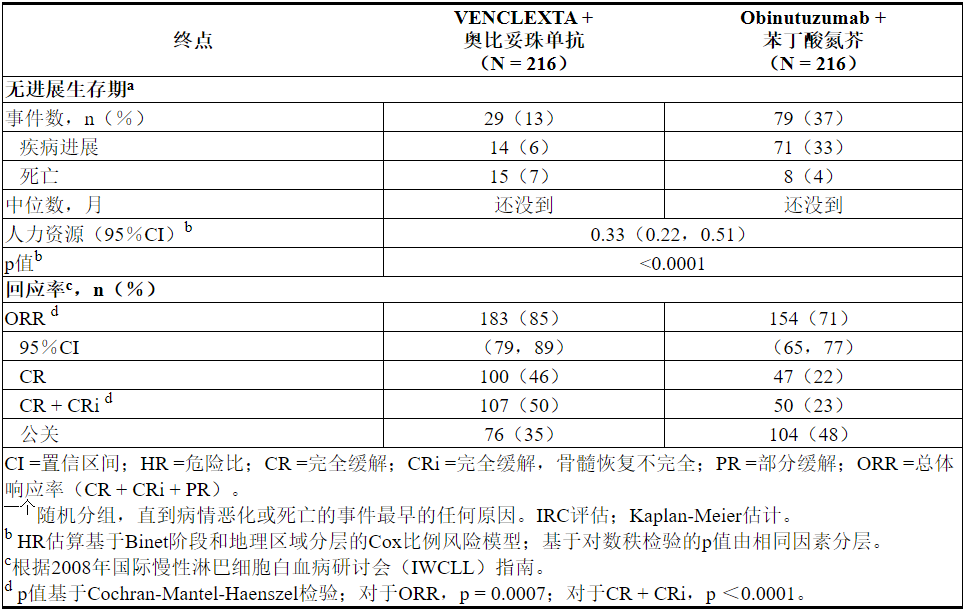
图1. CLL14中IRC评估的无进展生存期的Kaplan-Meier曲线
在分析时,尚未达到中位总体生存期(OS),只有不到10%的患者经历了这一事件。OS的中位随访时间为28个月。
使用等位基因特异性寡核苷酸聚合酶链反应(ASO-PCR)评估了最小残留疾病(MRD)。阴性状态的定义是每10 4白细胞少于1个CLL细胞。表20显示了治疗结束后3个月MRD阴性率(无论反应如何)和CR患者的情况。在该评估中,VEN + G组的134名外周血MRD阴性的患者匹配了骨髓标本。其中122名患者(91%)的外周血和骨髓MRD阴性。
表20.完成CLL14治疗后三个月的最小残留疾病阴性率
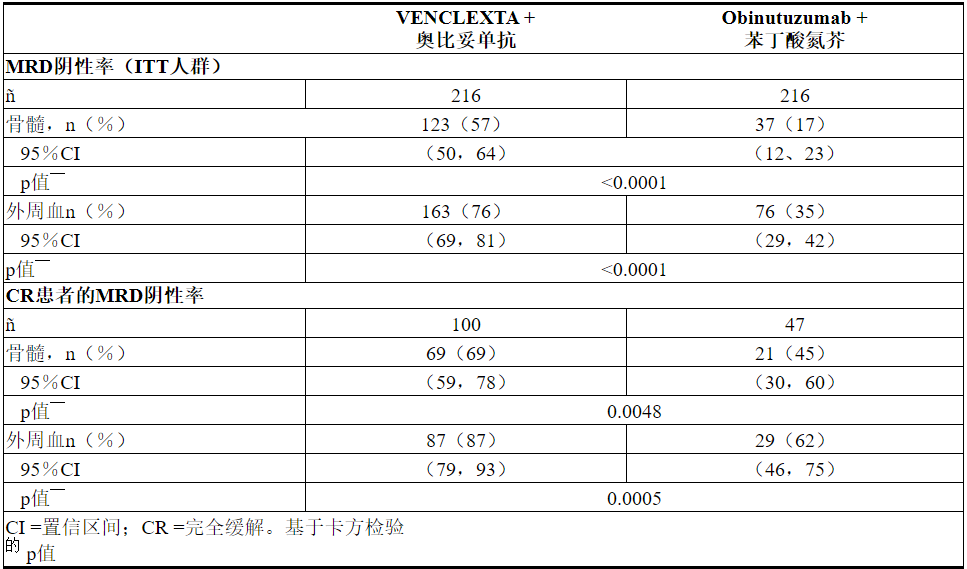
治疗结束后十二个月,VEN + G治疗的患者外周血MRD阴性率为58%(126/216),GClb治疗的患者为9%(20/216)。
村野
MURANO是一项随机(1:1),多中心,开放标签研究(NCT02005471),用于评估VENCLEXTA联合利妥昔单抗(VEN + R)与苯达莫司汀联合利妥昔单抗(B + R)在CLL患者中的疗效和安全性谁曾接受过至少一线的先前治疗。在VEN + R患者手臂完成了5周的斜升时间表[见剂量和给药(2.1,2.2)]和接收VENCLEXTA 400毫克,每天一次,从利妥昔单抗的周期1第1天24个月没有疾病进展的或不可接受的毒性。在第1个周期的第1天和500 mg / m 2的剂量增加5周后,以375 mg / m 2的剂量增加静脉注射利妥昔单抗在第2-6个周期的第1天。每个周期为28天。随机分组接受B + R治疗的患者在第1天和第2天以70 mg / m 2的剂量接受苯达莫司汀治疗6个周期(28天周期),并按上述剂量和时间表接受利妥昔单抗治疗。
总共389例患者被随机分组:VEN + R组为194名,B + R组为195名。VEN + R和B + R组的基线人口统计学和疾病特征相似。中位年龄为65岁(范围:22至85岁),白人为97%,男性为74%,ECOG行为状态<2为99%。先前治疗的中位数为1(范围:1至5);59%的患者接受过1种先前的治疗,26%的患者接受过2种先前的治疗,16%的患者接受过3种以上治疗。先前的疗法包括烷化剂(94%),抗CD20抗体(77%),B细胞受体途径抑制剂(2%)和先前的嘌呤类似物(81%,包括55%的氟达拉滨/环磷酰胺/利妥昔单抗)。在24%的患者中检测到17p缺失,在25%的患者中检测到TP53突变,在32%的患者中检测到11q缺失,并且IgVH未突变 在63%。
疗效基于IRC评估的PFS。PFS的中位随访时间为23.4个月(范围:0至37.4+个月)。
MURANO的功效结果示于表21中。PFS的Kaplan-Meier曲线如图2所示。
表21. MURANO中IRC评估的疗效结果
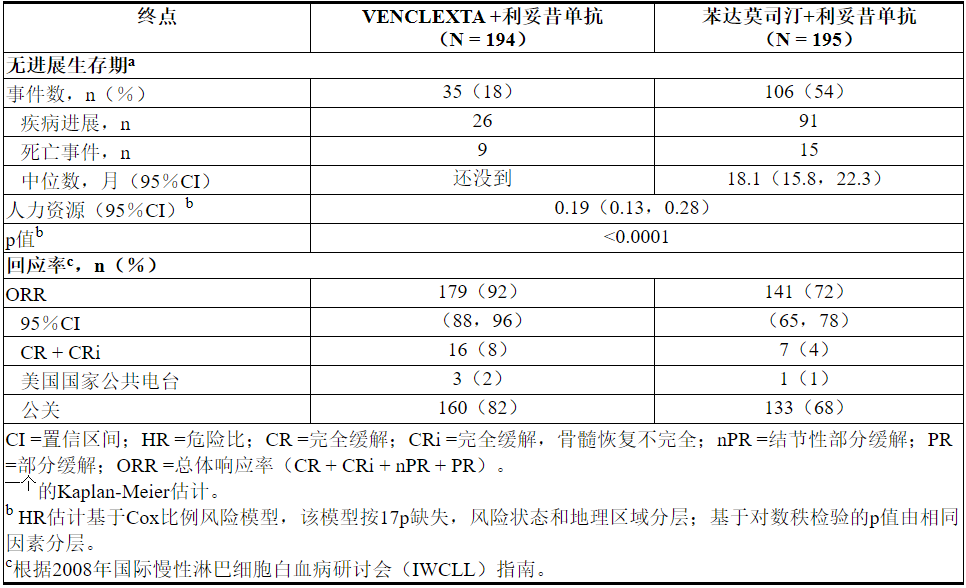
图2. MURANO中IRC评估的无进展生存期的Kaplan-Meier曲线
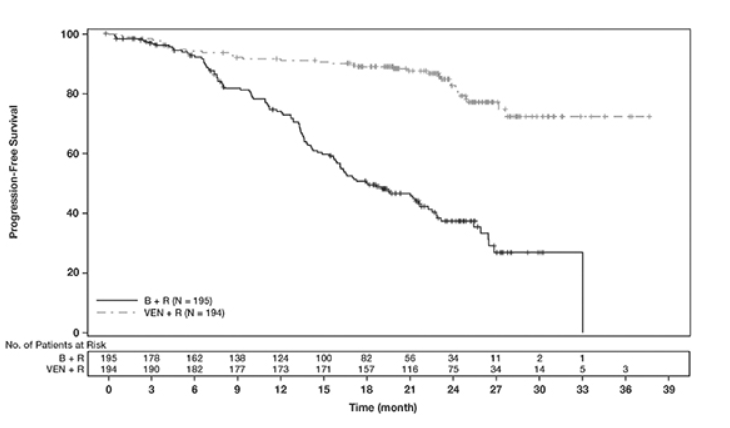
在分析时,中位随访22.9个月后,任一组均未达到中位总生存期。
在最后一次利妥昔单抗给药后3个月,达到PR或更高PR的患者外周血MRD阴性率为VEN + R组为53%(103/194),B +组为12%(23/195) R臂。在此时间点,VEN + R组的MRD阴性CR / CRi率为3%(6/194),B + R组为2%(3/195)。
单一疗法
VENCLEXTA单一疗法在先前治疗的CLL或SLL中的疗效基于三项单臂研究。
研究M13-982
VENCLEXTA的疗效已在研究M13-982(NCT01889186)中确立,M13-982是一项开放标签,单臂,多中心的临床试验,涉及106例CLP缺失17p缺失且至少接受过一种治疗的患者。在这项研究中,使用Vysis CLL FISH探针试剂盒确认了患者外周血标本中的17p缺失,该试剂盒已获得FDA批准,可以选择VENCLEXTA治疗的患者。患者通过每周递增的方案接受VENCLEXTA,起始剂量为20 mg,然后逐渐增加至50 mg,100 mg,200 mg,最后每天400 mg。患者继续每天一次口服400毫克VENCLEXTA,直到疾病进展或出现不可接受的毒性。
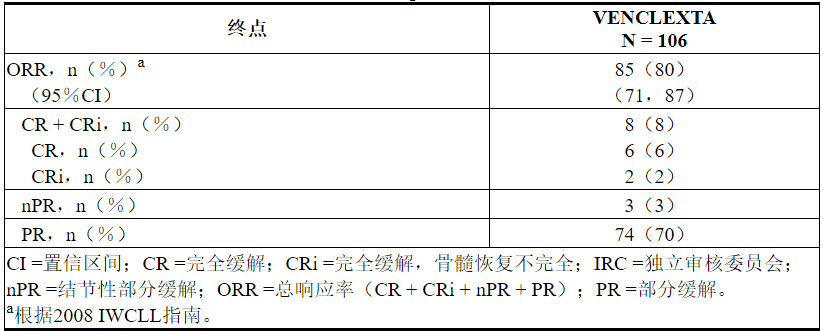
疗效基于IRC评估的总体缓解率(ORR)。
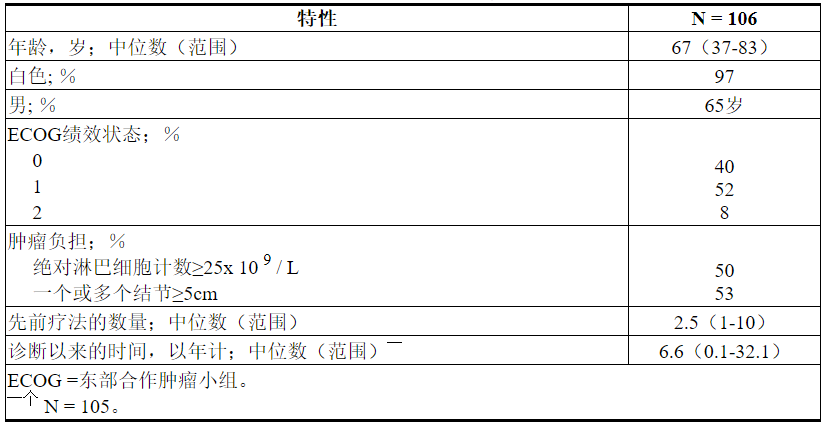
表22总结了研究人群的基线人口统计学和疾病特征。
表22.研究M13-982中的基线患者特征
评估时的中位治疗时间为12.1个月(范围:0至21.5个月)。功效结果示于表23。
表23.研究M13-982中先前治疗CLL 17p缺失的CLL患者每个IRC的疗效结果
首次反应的中位时间为0.8个月(范围:0.1到8.1个月)。
根据随后的数据截止日期和研究者评估的疗效,缓解持续时间(DOR)为2.9至32.8+个月。中位随访22个月未达到中位DOR。
在接受VENCLEXTA治疗后,对达到CR或CRi的患者的外周血和骨髓中的最小残留疾病进行了评估。3%(3/106)在外周血和骨髓中达到MRD阴性(每10 4个白细胞少于1个CLL细胞)。
研究M12-175
研究M12-175(NCT01328626)是一项多中心开放标签试验,纳入了先前治疗过的CLL或SLL患者,包括17p缺失患者。评估了每天接受400 mg VENCLEXTA的67例患者(其中CLL患者59例,SLL患者8例)的疗效。患者继续服用该剂量直至疾病进展或出现不可接受的毒性。评估时的中位治疗时间为22.1个月(范围:0.5至71.7个月)。
中位年龄为65岁(范围:42至84岁),男性占78%,白人占87%。先前治疗的中位数为3(范围:1至11)。在基线时,67%的患者有一个或多个结点≥5 cm,30%的患者ALC≥25 x 10 9 / L,33%的患者记录了未突变的IgVH,21%的患者记录了17p缺失。
根据2008 IWCLL指南评估疗效,并由IRC进行评估。ORR为76%(95%CI:64%,86%),CR + CRi率为10%,PR率为66%。DOR中位数为36.2个月(范围:2.4至52.4个月)。
研究M14-032
研究M14-032(NCT02141282)是一项开放标签,多中心研究,评估了VENCLEXTA在先前曾接受依鲁替尼或依达拉西治疗或进展的CLL患者中的疗效。在开始治疗后,患者每天接受400 mg VENCLEXTA的剂量。患者继续每天服用一次VENCLEXTA 400 mg,直到疾病进展或出现不可接受的毒性。在分析时,中位治疗时间为19.5个月(范围:0.1至39.5个月)。
在接受治疗的127例患者中(91例曾接受过ibrutinib,36例曾接受过idelalisib),中位年龄为66岁(范围:28至85岁),其中男性占70%,白人占92%。先前治疗的中位数为4(范围:1至15)。在基线时,41%的患者有一个或多个结点≥5 cm,31%的绝对淋巴细胞计数≥25x 10 9 / L,57 %的患者记录了未变异的IgVH,39%的患者记录了17p缺失。
功效基于2008 IWCLL指南,并由IRC评估。ORR为70%(95%CI:61%,78%),CR + CRi率为5%,PR率为65%。未达到中位DOR,中位随访时间为19.9个月(范围:2.9至36个月)。
14.2急性髓细胞白血病
VENCLEXTA在两项新诊断为≥75岁的AML或合并症的患者中至少根据以下标准之一进行了两项开放标签的非随机试验研究:合并EORG表现为2-3,严重的心脏或肺部合并症,中度肝功能不全,CLcr <45 mL / min或其他合并症。根据完全缓解率和持续时间确定疗效。
研究M14-358
在新诊断的AML患者中,VENCLEXTA与阿扎胞苷(N = 84)或地西他滨(N = 31)联合使用在VENCLEXTA的非随机,开放标签临床试验(NCT02203773)中进行了研究。在这些患者中,接受阿扎胞苷联合治疗的67例患者和接受地西他滨联合治疗的13例年龄在75岁或75岁以上,或患有合并症,无法使用强化诱导化疗。
患者通过每天一次最终剂量的最终剂量400 mg服用VENCLEXTA [参见剂量和用法(2.1)]。在启动期间,患者接受了TLS预防,并入院进行监测。阿扎胞苷75毫克/米2,在20任一施用静脉内或皮下对天每28天周期开始于第1周期第1天的地西他滨1-7毫克/米2从第1个周期的第1天开始,在每个28天周期的第1-5天静脉注射。患者继续接受治疗,直到疾病进展或出现不可接受的毒性。在临床试验中已实施了降低阿扎胞苷剂量以治疗血液学毒性的方法,请参阅阿扎胞苷的完整处方信息。在临床试验中未降低地西他滨的剂量。
表24总结了研究人群的基线人口统计学和疾病特征。
表24. VENCLEXTA与阿扎胞苷或地西他滨联合治疗的AML患者的基线患者特征
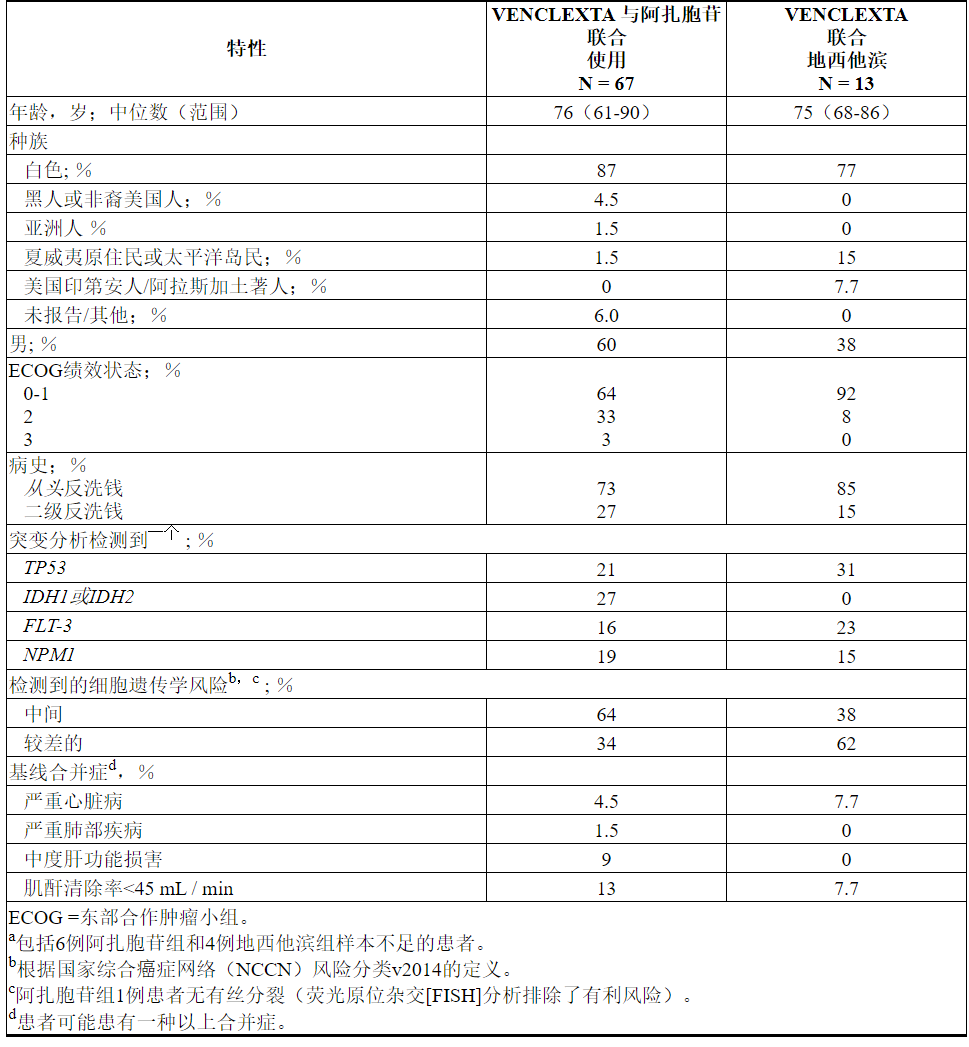
功效结果示于表25。
表25. VENCLEXTA与阿扎胞苷或地西他滨联用治疗的新诊断AML患者的疗效结果
VENCLEXTA联合阿扎胞苷的中位随访时间为7.9个月(0.4至36个月)。在分析时,对于获得CR的患者,观察到的缓解中位时间为5.5个月(范围:0.4到30个月)。观察到的缓解时间是从CR开始到数据截止日期或从CR复发的时间。
VENCLEXTA联合地西他滨的中位随访时间为11个月(0.7至21个月)。在分析时,对于获得CR的患者,观察到的缓解中位时间为4.7个月(范围:1.0到18个月)。观察到的缓解时间是从CR开始到数据截止日期或从CR复发的时间。
VENCLEXTA联合阿扎胞苷治疗的患者首次CR或CRh的中位时间为1.0个月(范围:0.7到8.9个月)。
VENCLEXTA联合地西他滨治疗的患者首次CR或CRh的中位时间为1.9个月(范围:0.8到4.2个月)。
在接受VENCLEXTA联合阿扎胞苷治疗的患者中,有7.5%(5/67)随后接受了干细胞移植。
该研究招募了另外35名患者(年龄范围:65至74岁),这些患者没有已知的合并症,无法使用强化诱导化疗,并接受VENCLEXTA联合阿扎胞苷(N = 17)或地西他滨(N = 18)治疗。
对于接受VENCLEXTA联合阿扎胞苷治疗的17例患者,CR率为35%(95%CI:14%,62%)。CRh率为41%(95%CI:18%,67%)。随后有七名(41%)患者接受了干细胞移植。
对于VENCLEXTA联合地西他滨治疗的18例患者,CR率为56%(95%CI:31%,79%)。CRh率为22%(95%CI:6.4%,48%)。随后有三名(17%)患者接受了干细胞移植。
研究M14-387
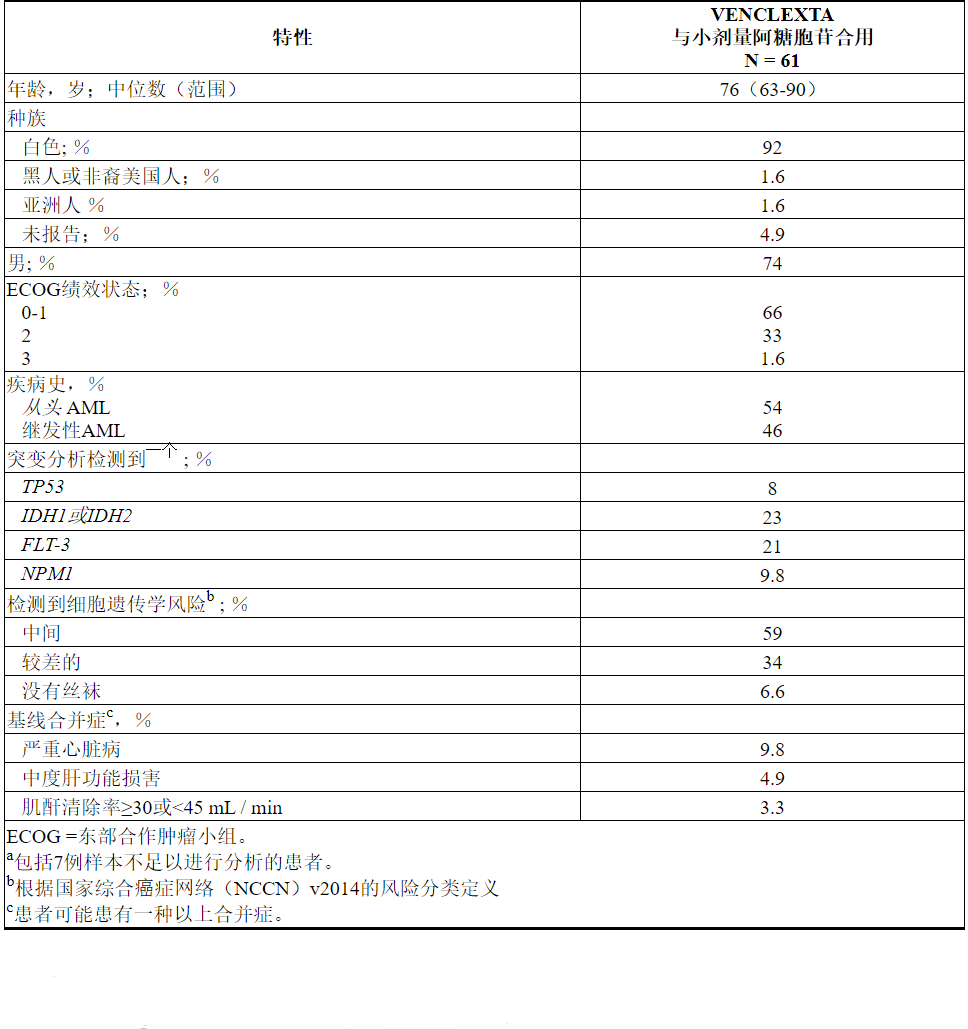
VENCLEXTA在VENCLEXTA与低剂量阿糖胞苷(N = 82)联合使用的非随机,开放标签临床试验(NCT02287233)中,用于新诊断为AML的患者,包括先前曾接受过低甲基化剂治疗的患者血液系统疾病。在这些患者中,有61名年龄在75岁或75岁以上,或患有合并症,因此至少根据以下标准之一使用强化诱导化疗:基线ECOG表现为2-3,严重的心脏或肺部合并症,中度肝损伤,CLcr≥30至<45 mL / min或其他合并症。
患者通过每日增加一次剂量最终一次每日一次最终剂量600 mg来启动VENCLEXTA [请参阅剂量和给药方法(2.1)]。在启动期间,患者接受了TLS预防,并入院进行监测。阿糖胞苷,剂量20毫克/米的2是每日一次皮下施用对1-10天每28天周期开始于1周期第1天的患者继续接受治疗周期,直至疾病进展或不可接受的毒性。在临床试验中未实施小剂量阿糖胞苷的减量。
表26总结了研究人群的基线人口统计学和疾病特征。
表26. VENCLEXTA与小剂量阿糖胞苷联合治疗的AML患者的基线患者特征
功效结果示于表27。
表27. VENCLEXTA与小剂量阿糖胞苷联合治疗的新诊断AML患者的疗效结果
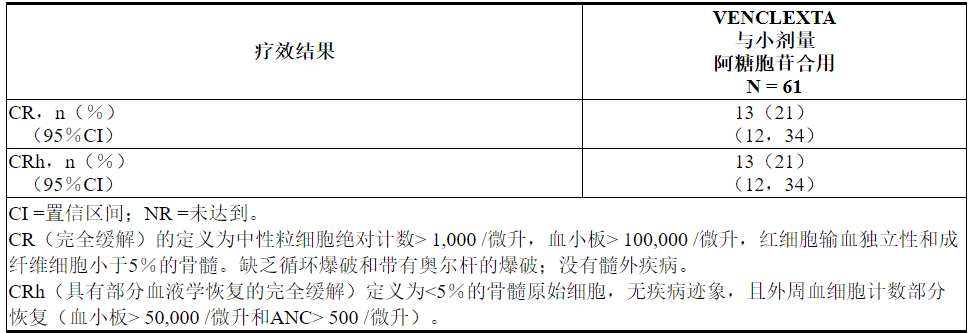
中位随访时间为6.5个月(范围:0.3到34个月)。在分析时,对于获得CR的患者,观察到的缓解中位时间中位数为6.0个月(范围:0.03至25个月)。观察到的缓解时间是从CR开始到数据截止日期或从CR复发的时间。
VENCLEXTA联合小剂量阿糖胞苷治疗的患者首次CR或CRh的中位时间为1.0个月(范围:0.8到9.4个月)。
该研究招募了另外21名患者(年龄范围:67至74岁),这些患者没有已知的合并症,不能使用强化诱导化疗,并接受VENCLEXTA联合小剂量阿糖胞苷治疗。CR率为33%(95%CI:15%,57%)。CRh率为24%(95%CI:8.2%,47%)。一名患者(4.8%)随后接受了干细胞移植。
16供应/存储和处理方式
VENCLEXTA的分配如下:

VENCLEXTA 10 mg薄膜衣片为圆形,双凸形,浅黄色,在一侧凹有“ V”,在另一侧凹有“ 10”。
VENCLEXTA 50 mg薄膜衣片为长方形,双凸形,米色,凹陷处一侧带有“ V”,另一侧带有“ 50”。
VENCLEXTA 100 mg薄膜衣片为长方形,双凸形,浅黄色,在一侧凹有“ V”,在另一侧凹有“ 100”。
存放在或低于86°F(30°C)。
17患者咨询信息
建议患者阅读FDA批准的患者标签(用药指南)。
- 肿瘤溶解综合征
建议患者TLS的潜在风险,尤其是在治疗开始时和加速阶段期间,并立即报告与此事件相关的任何体征和症状(发烧,发冷,恶心,呕吐,意识模糊,呼吸急促,癫痫发作,心律不齐,尿液发黑或浑浊,异常疲倦,肌肉疼痛和/或关节不适),请其医疗保健提供者(HCP)进行评估[请参阅警告和注意事项(5.1)]。
建议服用VENCLEXTA的患者每天补充水分,以减少TLS的风险。推荐的水量是每天6到8杯水(总计约56盎司)。患者应在第一剂开始前2天和当天开始喝水,每次增加剂量时都应喝水[见剂量和给药方法(2.2) ]。
告知患者定期安排血液检查或其他实验室检查的重要性[参见剂量和用法(2.2)]。
告知患者可能有必要在医院或医疗办公室使用VENCLEXTA,以进行TLS监测。 - 中性粒细胞减少
建议患者如果发烧或有任何感染迹象,请立即联系他们的HCP。告知患者需要定期监测血球计数[见警告和注意事项(5.2)]。 - 感染
建议患者如果发烧或有任何感染迹象,请立即联系他们的HCP [请参阅警告和注意事项(5.3)]。 - 药物相互作用
建议患者在使用VENCLEXTA治疗期间避免食用柚子产品,塞维利亚橘子或杨桃。告知患者VENCLEXTA可能与某些药物相互作用;因此,建议患者将任何处方药,非处方药,维生素和草药产品的使用情况告知其医疗保健提供者[请参阅禁忌症(4)和药物相互作用(7.1)]。 - 免疫接种
建议患者避免接种活疫苗,因为在VENCLEXTA治疗期间它们可能不安全或无效[请参阅警告和注意事项(5.4)]。 - 怀孕和哺乳期
建议妇女在使用VENCLEXTA治疗期间可能对胎儿造成潜在危险,并避免怀孕。建议有生殖潜力的女性患者在治疗期间以及完成治疗后至少30天使用有效避孕方法。建议女性在接受VENCLEXTA治疗期间怀孕或怀疑怀孕时联系其HCP。还建议患者不进行母乳喂养同时服用VENCLEXTA [见警告和注意事项(5.5),和使用特殊人群中(8.1,8.2,以及8.3)]。 - 男性不育症
的不育和生殖潜在的男性可能使用精子银行的可能性,建议患者[见特殊人群中使用(8.3)]。
服用VENCLEXTA的说明
建议患者严格按照处方服用VENCLEXTA,除非他们的HCP告知他们不要改变剂量或停止服用VENCLEXTA。建议患者根据HCP的指示,每天大约在同一时间口服一次VENCLEXTA,并应随餐和饮水一起吞服药片,不要被咀嚼,压碎或弄碎[参见剂量和用法(2.1)]。
建议有CLL / SLL的患者在治疗的前4周内将VENCLEXTA保留在原始包装中,并且不要将片剂转移到其他容器中。
劝告患者,如果在少于8小时的时间内错过了VENCLEXTA剂量,请立即服用错过的剂量并照常服用下一次剂量。如果错过VENCLEXTA剂量超过8小时,建议患者在平时等待并服用下一个剂量[见剂量和用法(2.6)]。
劝告患者在服用VENCLEXTA后呕吐的那一天不要再服用任何剂量,并在第二天的通常时间服用下一剂。
生产和销售:
AbbVie Inc.
北芝加哥,伊利诺伊州60064和
销售商:
Genentech USA,Inc
.罗氏集团成员,
南旧金山,加利福尼亚州94080-4990©2019 AbbVie Inc.
©2019 Genentech,
Inc.03-B947 2019年7月




NDC 0074-0579-28
CLL / SLL入门包
VENCLEXTA®
(venetoclax片)
10 mg,50 mg和100 mg
起始包
!警告
当您收到这种药物时,请与您的医生联系。
可能有必要在医生在场的情况下服用第一剂,以防止潜在的严重副作用。
分配器:每次分配VENCLEXTA时,请给患者附上随附的用药指南。
阿比
仅Rx
基因技术
NDC 0074–0561–14
仅Rx
VENCLEXTA®
(venetoclax片)
10毫克
14片
将随附的用药指南分配给每个患者。
阿比
基因技术

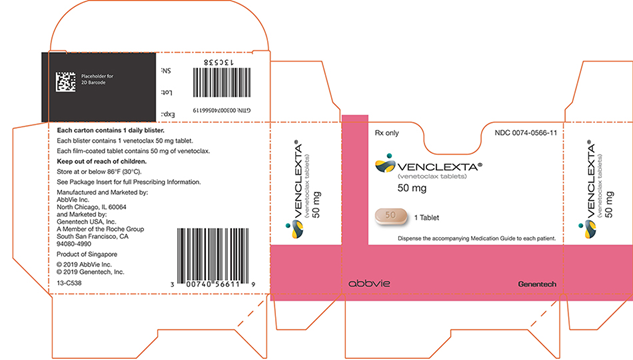
NDC 0074-0566-11
仅Rx
VENCLEXTA®
(venetoclax片)
50毫克
1片
将随附的用药指南分配给每个患者。
阿比
基因技术
NDC 0074-0576-22
仅Rx
VENCLEXTA®
(venetoclax片)
100毫克
120片
如果瓶口的密封件损坏或丢失,请勿接受。
每片薄膜包衣的片剂含有100毫克的Venetoclax。
请将本品放在儿童不能接触的地方。
存放在或低于86°F(30°C)。
阿比
基因技术

NDC 0074-0576-22
仅Rx
VENCLEXTA®
(venetoclax片)
100毫克
120片
将随附的用药指南分配给每个患者。
阿比
基因技术

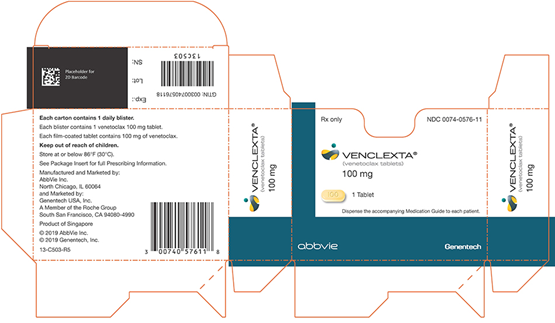
NDC 0074–0576–11
仅Rx
VENCLEXTA®
(venetoclax片)
100毫克
1片
将随附的用药指南分配给每个患者。
阿比
基因技术
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。
-
本说明书来源于FDA网站
https://nctr-crs.fda.gov/fdalabel/services/spl/set-ids/b118a40d-6b56-cee3-10f6-ded821a97018/spl-doc?hl=venetoclax
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Chronic Lymphocytic Leukemia/Small Lymphocytic LymphomaVENCLEXTA is indicated for the treatment of adult patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL).
1.2 Acute Myeloid Leukemia
VENCLEXTA is indicated in combination with azacitidine, or decitabine, or low-dose cytarabine for the treatment of newly-diagnosed acute myeloid leukemia (AML) in adults who are age 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy.
This indication is approved under accelerated approval based on response rates [see Clinical Studies (14.2)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Assess patient-specific factors for level of risk of tumor lysis syndrome (TLS) and provide prophylactic hydration and anti-hyperuricemics to patients prior to first dose of VENCLEXTA to reduce risk of TLS [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
Instruct patients to take VENCLEXTA tablets with a meal and water at approximately the same time each day. VENCLEXTA tablets should be swallowed whole and not chewed, crushed, or broken prior to swallowing.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
VENCLEXTA dosing begins with a 5-week ramp-up.
VENCLEXTA 5-week Dose Ramp-Up Schedule
Administer the VENCLEXTA dose according to a weekly ramp-up schedule over 5 weeks to the recommended daily dose of 400 mg as shown in Table 1. The 5-week ramp-up dosing schedule is designed to gradually reduce tumor burden (debulk) and decrease the risk of TLS.
Table 1. Dosing Schedule for Ramp-Up Phase in Patients with CLL/SLL
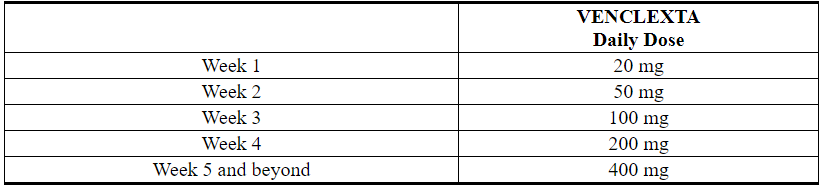
The CLL/SLL Starting Pack provides the first 4 weeks of VENCLEXTA according to the ramp-up schedule. The 400 mg dose is achieved using 100 mg tablets supplied in bottles [see How Supplied/Storage and Handling (16)].
VENCLEXTA in Combination with Obinutuzumab
Start obinutuzumab administration at 100 mg on Cycle 1 Day 1, followed by 900 mg on Cycle 1 Day 2. Administer 1000 mg on Days 8 and 15 of Cycle 1 and on Day 1 of each subsequent 28-day cycle, for a total of 6 cycles. Refer to the obinutuzumab prescribing information for recommended obinutuzumab dosing information.
On Cycle 1 Day 22, start VENCLEXTA according to the 5-week ramp-up schedule (see Table 1). After completing the ramp-up schedule on Cycle 2 Day 28, patients should continue VENCLEXTA 400 mg once daily from Cycle 3 Day 1 until the last day of Cycle 12.
VENCLEXTA in Combination with Rituximab
Start rituximab administration after the patient has completed the 5-week dose ramp-up schedule with VENCLEXTA (see Table 1) and has received the 400 mg dose of VENCLEXTA for 7 days. Administer rituximab on Day 1 of each 28-day cycle for 6 cycles, with rituximab dosed at 375 mg/m2 intravenously for Cycle 1 and 500 mg/m2 intravenously for Cycles 2-6.
Patients should continue VENCLEXTA 400 mg once daily for 24 months from Cycle 1 Day 1 of rituximab.
VENCLEXTA as Monotherapy
The recommended dose of VENCLEXTA is 400 mg once daily after the patient has completed the 5-week dose ramp-up schedule. VENCLEXTA should be taken orally once daily until disease progression or unacceptable toxicity is observed.
Acute Myeloid Leukemia
The dose of VENCLEXTA depends upon the combination agent.
The VENCLEXTA dosing schedule (including ramp-up) is shown in Table 2. Initiate the azacitidine or decitabine or low-dose cytarabine on Day 1.
Table 2. Dosing Schedule for Ramp-up Phase in Patients with AML
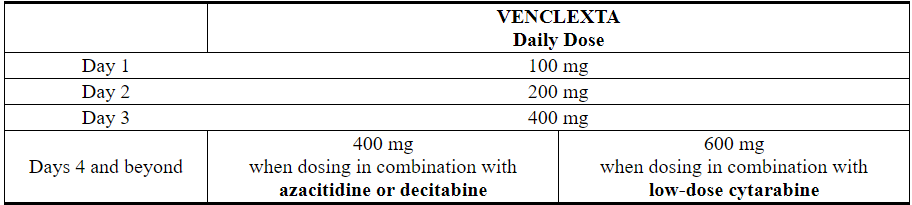
Continue VENCLEXTA, in combination with azacitidine or decitabine or low-dose cytarabine, until disease progression or unacceptable toxicity is observed.
2.2 Risk Assessment and Prophylaxis for Tumor Lysis Syndrome
Patients treated with VENCLEXTA may develop tumor lysis syndrome. Refer to the appropriate section below for specific details on management. Assess patient-specific factors for level of risk of tumor lysis syndrome (TLS) and provide prophylactic hydration and anti-hyperuricemics to patients prior to first dose of VENCLEXTA to reduce risk of TLS.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
VENCLEXTA can cause rapid reduction in tumor and thus poses a risk for TLS in the initial 5-week ramp-up phase. Changes in blood chemistries consistent with TLS that require prompt management can occur as early as 6 to 8 hours following the first dose of VENCLEXTA and at each dose increase.
The risk of TLS is a continuum based on multiple factors, including tumor burden and comorbidities. Reduced renal function (creatinine clearance [CLcr] <80 mL/min) further increases the risk. Perform tumor burden assessments, including radiographic evaluation (e.g., CT scan), assess blood chemistry (potassium, uric acid, phosphorus, calcium, and creatinine) in all patients and correct pre-existing abnormalities prior to initiation of treatment with VENCLEXTA. The risk may decrease as tumor burden decreases [see Warnings and Precautions (5.1) and Use in Specific Populations (8.6)].
Table 3 below describes the recommended TLS prophylaxis and monitoring during VENCLEXTA treatment based on tumor burden determination from clinical trial data. Consider all patient comorbidities before final determination of prophylaxis and monitoring schedule.
Table 3. Recommended TLS Prophylaxis Based on Tumor Burden in Patients with CLL/SLL
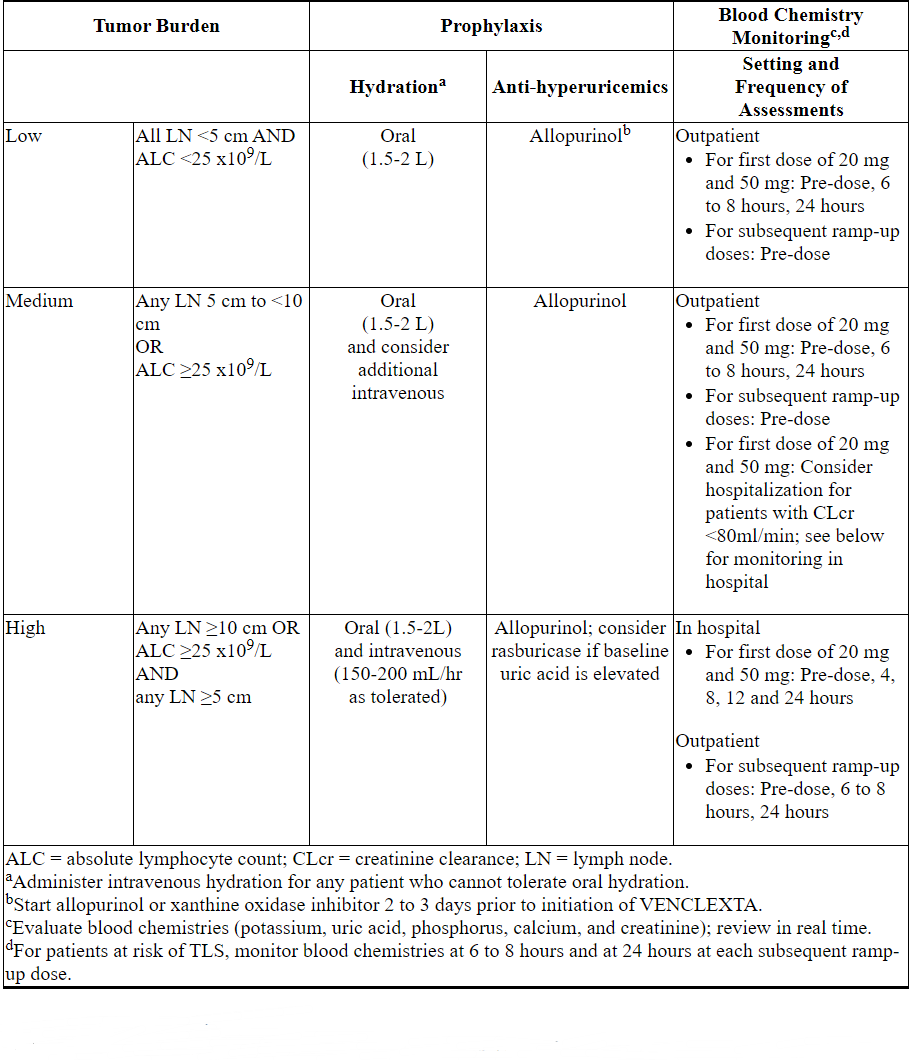
Acute Myeloid Leukemia
- All patients should have white blood cell count less than 25 × 109/L prior to initiation of VENCLEXTA. Cytoreduction prior to treatment may be required.
- Prior to first VENCLEXTA dose, provide all patients with prophylactic measures including adequate hydration and anti-hyperuricemic agents and continue during ramp-up phase.
- Assess blood chemistry (potassium, uric acid, phosphorus, calcium, and creatinine) and correct pre-existing abnormalities prior to initiation of treatment with VENCLEXTA.
- Monitor blood chemistries for TLS at pre-dose, 6 to 8 hours after each new dose during ramp-up and 24 hours after reaching final dose.
- For patients with risk factors for TLS (e.g., circulating blasts, high burden of leukemia involvement in bone marrow, elevated pretreatment lactate dehydrogenase (LDH) levels, or reduced renal function) additional measures should be considered, including increased laboratory monitoring and reducing VENCLEXTA starting dose.
2.3 Dosage Modifications Based on Toxicities
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
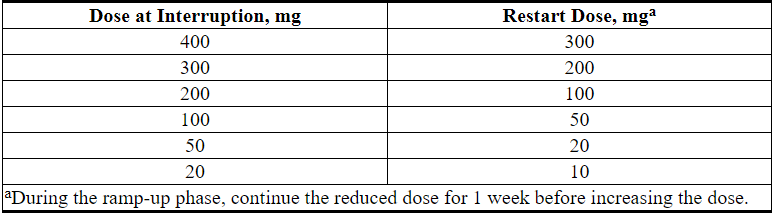
Interrupt dosing or reduce dose for toxicities. See Table 4 and Table 5 for recommended dose modifications for toxicities related to VENCLEXTA. For patients who have had a dosing interruption greater than 1 week during the first 5 weeks of ramp-up phase or greater than 2 weeks after completing the ramp-up phase, reassess for risk of TLS to determine if reinitiation with a reduced dose is necessary (e.g., all or some levels of the dose ramp-up schedule) [see Dosage and Administration (2.1, 2.2)].
Table 4. Recommended VENCLEXTA Dose Modifications for Toxicitiesa in CLL/SLL
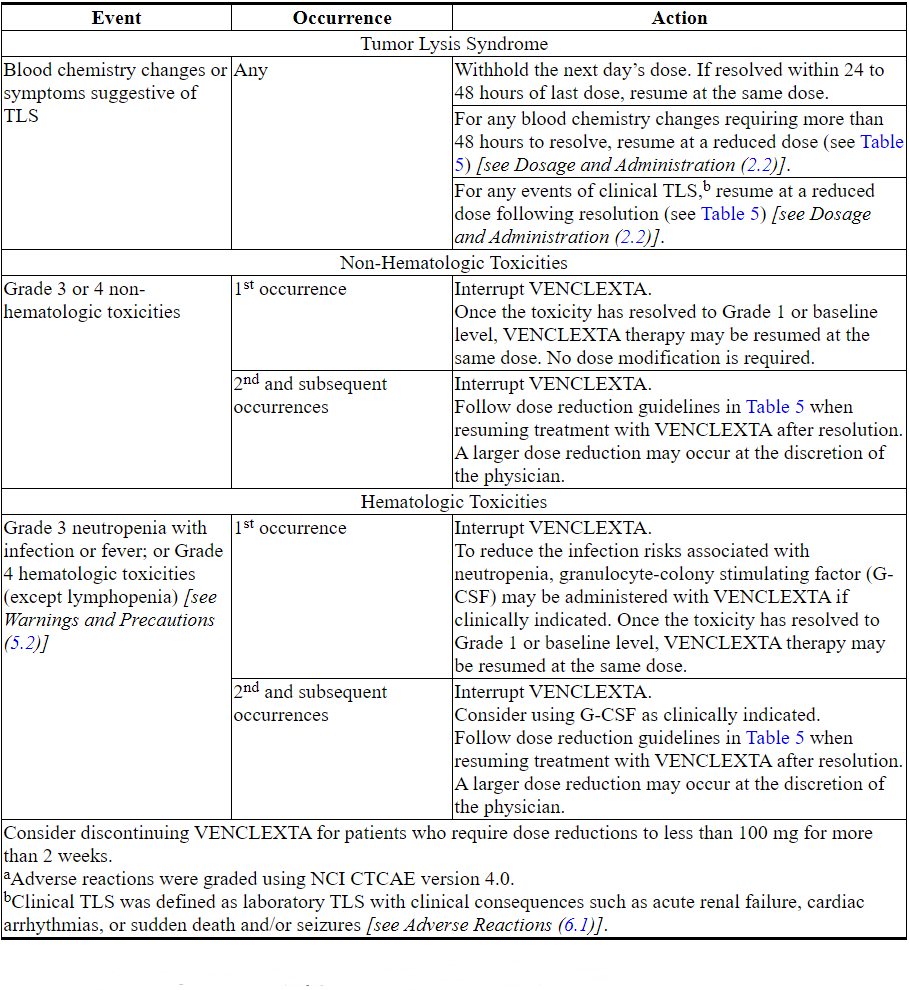
Table 5. Dose Reduction for Toxicity During VENCLEXTA Treatment in CLL/SLL
Acute Myeloid Leukemia
Monitor blood counts frequently through resolution of cytopenias. Management of some adverse reactions [see Warnings and Precautions (5.2) and Adverse Reactions (6.2)] may require dose interruptions or permanent discontinuation of VENCLEXTA. Table 6 shows the dose modification guidelines for hematologic toxicities.
Table 6. Recommended Dose Modifications for Toxicitiesa in AML
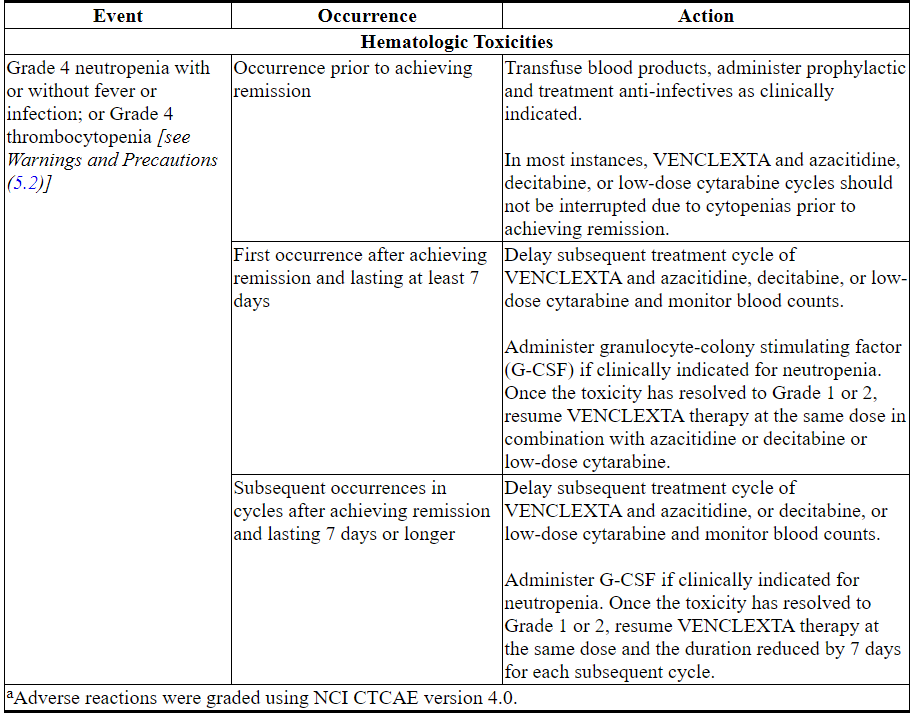
2.4 Dosage Modifications for Concomitant Use with Strong or Moderate CYP3A Inhibitors or P-gp Inhibitors
Table 7 describes VENCLEXTA contraindication or dosage modification based on concomitant use with a strong or moderate CYP3A inhibitor or P-gp inhibitor [see Drug Interactions (7.1)] at initiation, during, or after the ramp-up phase.
Resume the VENCLEXTA dosage that was used prior to concomitant use of a strong or moderate CYP3A inhibitor or P-gp inhibitor 2 to 3 days after discontinuation of the inhibitor [see Dosage and Administration (2.3) and Drug Interactions (7.1)].
Table 7. Management of Potential VENCLEXTA Interactions with CYP3A and P-gp Inhibitors
2.5 Dosage Modifications for Patients with Severe Hepatic Impairment
Reduce the VENCLEXTA once daily dose by 50% for patients with severe hepatic impairment (Child-Pugh C); monitor these patients more closely for signs of toxicity [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
2.6 Missed Dose
If the patient misses a dose of VENCLEXTA within 8 hours of the time it is usually taken, the patient should take the missed dose as soon as possible and resume the normal daily dosing schedule. If a patient misses a dose by more than 8 hours, the patient should not take the missed dose and should resume the usual dosing schedule the next day.
If the patient vomits following dosing, no additional dose should be taken that day. The next prescribed dose should be taken at the usual time.
3 DOSAGE FORMS AND STRENGTHS
Table 8. VENCLEXTA Tablet Strength and Description

4 CONTRAINDICATIONS
Concomitant use of VENCLEXTA with strong CYP3A inhibitors at initiation and during the ramp-up phase is contraindicated in patients with CLL/SLL due to the potential for increased risk of tumor lysis syndrome [see Dosage and Administration (2.4) and Drug Interactions (7.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Tumor Lysis Syndrome
Tumor lysis syndrome (TLS), including fatal events and renal failure requiring dialysis, has occurred in patients with high tumor burden when treated with VENCLEXTA [see Adverse Reactions (6.1, 6.2)].
In patients with CLL who followed the current (5 week) dose ramp-up and the TLS prophylaxis and monitoring measures, the rate of TLS was 2% in the VENCLEXTA CLL monotherapy studies. The rate of TLS remained consistent with VENCLEXTA in combination with obinutuzumab or rituximab. With a 2 to 3 week dose ramp-up and higher starting dose in patients with CLL/SLL, the TLS rate was 13% and included deaths and renal failure [see Adverse Reactions (6.1)].
VENCLEXTA can cause rapid reduction in tumor and thus poses a risk for TLS at initiation and during the ramp-up phase. Changes in blood chemistries consistent with TLS that require prompt management can occur as early as 6 to 8 hours following the first dose of VENCLEXTA and at each dose increase.
The risk of TLS is a continuum based on multiple factors, including tumor burden and comorbidities. Reduced renal function further increases the risk. Patients should be assessed for risk and should receive appropriate prophylaxis for TLS, including hydration and anti-hyperuricemics. Monitor blood chemistries and manage abnormalities promptly. Interrupt dosing if needed. Employ more intensive measures (intravenous hydration, frequent monitoring, hospitalization) as overall risk increases [see Dosage and Administration (2.2, 2.3) and Use in Specific Populations (8.6)].
Concomitant use of VENCLEXTA with P-gp inhibitors or strong or moderate CYP3A inhibitors increases venetoclax exposure, may increase the risk of TLS at initiation and during ramp-up phase and requires VENCLEXTA dose adjustment [see Dosage and Administration (2.4) and Drug Interactions (7.1)].
5.2 Neutropenia
In patients with CLL, Grade 3 or 4 neutropenia developed in 63% to 64% of patients and Grade 4 neutropenia developed in 31% to 33% of patients treated with VENCLEXTA in combination and monotherapy studies (see Tables 10, 12, 14). Febrile neutropenia occurred in 4% to 6% of patients treated with VENCLEXTA in combination and monotherapy studies[see Adverse Reactions (6.1)].
In patients with AML, baseline neutrophil counts worsened in 97% to 100% of patients treated with VENCLEXTA in combination with azacitidine or decitabine or low-dose cytarabine. Neutropenia can recur with subsequent cycles of therapy.
Monitor complete blood counts throughout the treatment period. Interrupt dosing or reduce dose for severe neutropenia. Consider supportive measures including antimicrobials for signs of infection and use of growth factors (e.g., G-CSF) [see Dosage and Administration (2.3)].
5.3 Infections
Fatal and serious infections such as pneumonia and sepsis have occurred in patients treated with VENCLEXTA [see Adverse Reactions (6.1)]. Monitor patients closely for signs and symptoms of infection and treat promptly. Withhold VENCLEXTA for Grade 3 and higher infection [see Dosage and Administration (2.3)].
5.4 Immunization
Do not administer live attenuated vaccines prior to, during, or after treatment with VENCLEXTA until B-cell recovery occurs. The safety and efficacy of immunization with live attenuated vaccines during or following VENCLEXTA therapy have not been studied. Advise patients that vaccinations may be less effective.
5.5 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animals, VENCLEXTA may cause embryo-fetal harm when administered to a pregnant woman. In an embryo-fetal study conducted in mice, administration of venetoclax to pregnant animals at exposures equivalent to that observed in patients at a dose of 400 mg daily resulted in post-implantation loss and decreased fetal weight. There are no adequate and well-controlled studies in pregnant women using VENCLEXTA. Advise females of reproductive potential to avoid pregnancy during treatment. If VENCLEXTA is used during pregnancy or if the patient becomes pregnant while taking VENCLEXTA, the patient should be apprised of the potential hazard to the fetus [see Use in Specific Populations (8.1)].
5.6 Increased Mortality in Patients with Multiple Myeloma when VENCLEXTA is Added to Bortezomib and Dexamethasone
In a randomized trial (BELLINI; NCT02755597) in patients with relapsed or refractory multiple myeloma, the addition of VENCLEXTA to bortezomib plus dexamethasone, a use for which VENCLEXTA is not indicated, resulted in increased mortality. Treatment of patients with multiple myeloma with VENCLEXTA in combination with bortezomib plus dexamethasone is not recommended outside of controlled clinical trials.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Tumor Lysis Syndrome [see Warnings and Precautions (5.1)]
- Neutropenia [see Warnings and Precautions (5.2)]
- Infections [see Warnings and Precautions (5.3)]
Because clinical trials are conducted under widely variable conditions, adverse event rates observed in clinical trials of a drug cannot be directly compared with rates of clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trial Experience with Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
CLL14
The safety of VENCLEXTA in combination with obinutuzumab (VEN+G) versus obinutuzumab in combination with chlorambucil (GClb) was evaluated in a randomized, open-label, actively controlled trial in patients with previously untreated CLL.
Patients randomized to the VEN+G arm were treated with VENCLEXTA and obinutuzumab in combination for six cycles, then with VENCLEXTA as monotherapy for an additional six cycles. Patients initiated the first dose of the 5 week ramp-up for VENCLEXTA on Day 22 of Cycle 1 and once completed, continued VENCLEXTA 400 mg once daily for a total of 12 cycles. Details of the study treatment are described in Section 14 [see Clinical Studies (14.1)]. The trial required a total Cumulative Illness Rating Scale (CIRS) >6 or CLcr <70 mL/min, hepatic transaminases and total bilirubin ≤2 times upper limit of normal, and excluded patients with any individual organ/system impairment score of 4 by CIRS except eye, ear, nose, and throat organ system.
A total of 426 patients were treated (212 with VEN+G, 214 with GClb). The median duration of exposure to VENCLEXTA was 10.5 months (range: 0 to 13.5 months). The median number of cycles was 6 for obinutuzumab and 12 for chlorambucil.
In the VEN+G arm, fatal adverse reactions that occurred in the absence of disease progression and with onset within 28 days of the last study treatment were reported in 2% (4/212) of patients, most often from infection. Serious adverse reactions were reported in 49% of patients in the VEN+G arm, most often due to febrile neutropenia and pneumonia (5% each).
In the VEN+G arm, adverse reactions led to treatment discontinuation in 16% of patients, dose reduction in 21%, and dose interruption in 74%. In the VEN+G arm, neutropenia led to dose interruption of VENCLEXTA in 41% of patients, reduction in 13%, and discontinuation in 2%.
Table 9 and Table 10 present adverse reactions and laboratory abnormalities identified in the CLL14 trial, respectively. The most common (≥15%) adverse reactions observed with VEN+G were neutropenia, diarrhea, fatigue, nausea, anemia, and upper respiratory tract infection.
Table 9. Common (≥10%) Adverse Reactions in Patients Treated with VEN+G
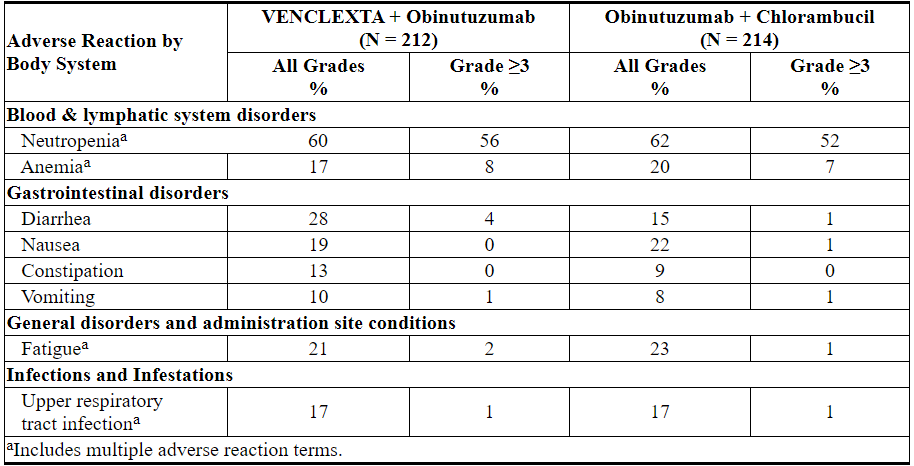
Other clinically important adverse reactions (all Grades) reported in <10% of patients treated with VEN+G are presented below:Blood & lymphatic system disorders: febrile neutropenia (6%)
Infection and infestations (all include multiple adverse reaction terms): pneumonia (9%), urinary tract infection (6%), sepsis (4%)
Metabolism and nutrition disorder: tumor lysis syndrome (1%)
During treatment with single agent VENCLEXTA after completion of VEN+G combination treatment, the most common all grade adverse reaction (≥10% patients) reported was neutropenia (26%). The most common grade ≥3 adverse reactions (≥2% patients) were neutropenia (23%), and anemia (2%).
Table 10. New or Worsening Clinically Important Laboratory Abnormalities Occurring at ≥10% in Patients Treated with VEN+G
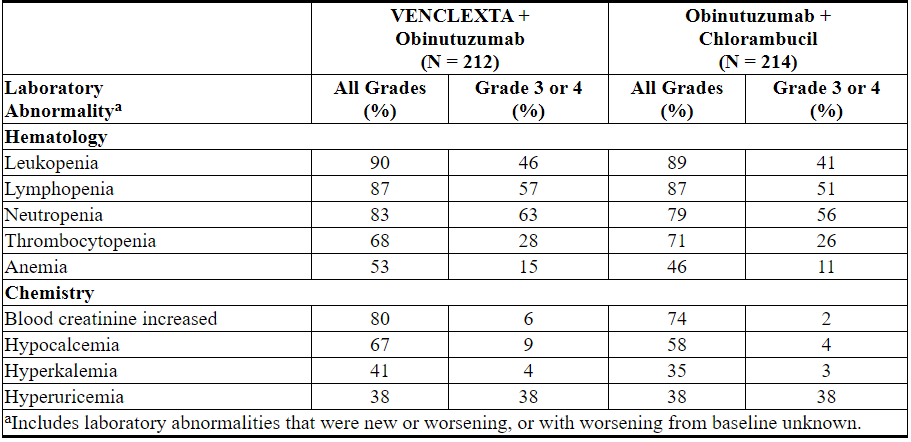
Grade 4 laboratory abnormalities developing in ≥2% of patients treated with VEN+G include neutropenia (32%), leukopenia and lymphopenia (10%), thrombocytopenia (8%), hypocalcemia (8%), hyperuricemia (7%), blood creatinine increased (3%), hypercalcemia (3%), and hypokalemia (2%).
MURANO
The safety of VENCLEXTA in combination with rituximab (VEN+R) versus bendamustine in combination with rituximab (B+R), was evaluated in an open-label randomized study, in patients with CLL who had received at least one prior therapy.
Patients randomized to VEN+R completed the scheduled ramp-up (5 weeks) and received VENCLEXTA 400 mg once daily in combination with rituximab for 6 cycles followed by single agent VENCLEXTA for a total of 24 months after ramp-up. Patients randomized to B+R received 6 cycles (28 days per cycle) for a total of 6 months. Details of the study treatment are described in Section 14 [see Clinical Studies (14.1)].
At the time of analysis, the median duration of exposure was 22 months in the VEN+R arm compared with 6 months in the B+R arm.
In the VEN+R arm, fatal adverse reactions that occurred in the absence of disease progression and within 30 days of the last VENCLEXTA treatment and/or 90 days of last rituximab were reported in 2% (4/194) of patients. Serious adverse reactions were reported in 46% of patients in the VEN+R arm, with most frequent (≥5%) being pneumonia (9%).
In the VEN+R arm, adverse reactions led to treatment discontinuation in 16% of patients, dose reduction in 15%, and dose interruption in 71%. In the B+R arm, adverse reactions led to treatment discontinuation in 10% of patients, dose reduction in 15%, and dose interruption in 40%. In the VEN+R arm, neutropenia led to dose interruption of VENCLEXTA in 46% of patients and discontinuation in 3%, and thrombocytopenia led to discontinuation in 3% of patients.
Table 11 and Table 12 present adverse reactions and laboratory abnormalities, respectively, identified in the MURANO trial. The MURANO trial was not designed to demonstrate a statistically significant difference in adverse reaction rates for VEN+R as compared with B+R, for any specific adverse reaction or laboratory abnormality.
Table 11. Common (≥10%) Adverse Reactions Reported with ≥5% Higher All-Grade or ≥2% Higher Grade ≥3 Incidence in Patients Treated with VEN+R Compared with B+R
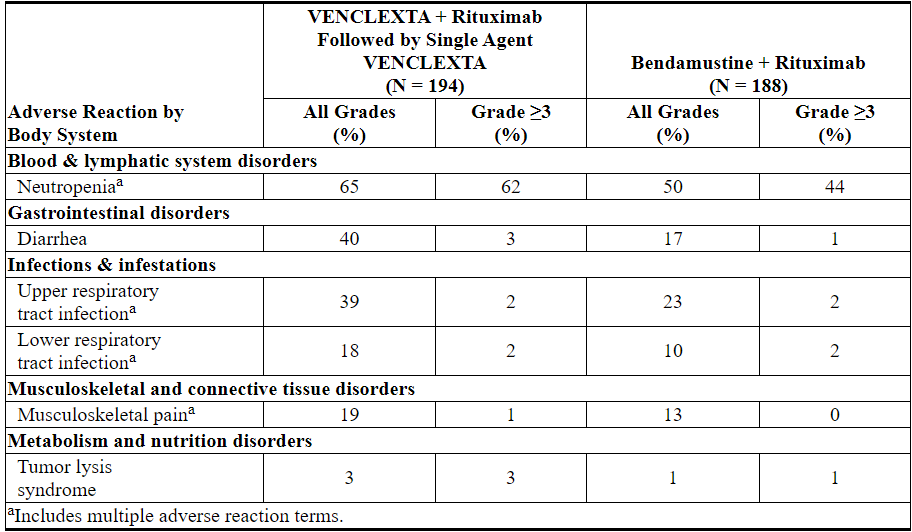
Other adverse reactions (all grades) reported in ≥10% of patients in the VEN+R arm in MURANO, and other important adverse reactions are presented below:
Blood & lymphatic system disorders: anemia (16%), thrombocytopenia (15%), febrile neutropenia (4%)
Gastrointestinal disorders: nausea (21%), constipation (14%), abdominal pain (13%), mucositis (10%), vomiting (8%)
Respiratory disorders: cough (22%)
General disorders and administration site conditions: fatigue (22%), pyrexia (15%)
Skin disorders: rash (13%)
Nervous system and psychiatric disorders: headache (11%), insomnia (11%)
Infections & infestations: pneumonia (10%), sepsis (1%)
During treatment with single agent VENCLEXTA after completion of VEN+R combination treatment, the most common all grade adverse reactions (≥10% patients) reported were upper respiratory tract infection (21%), diarrhea (19%), neutropenia (16%), and lower respiratory tract infections (11%). The most common Grade 3 or 4 adverse reactions (≥2% patients) were neutropenia (12%) and anemia (3%).
Laboratory Abnormalities
Table 12 describes common treatment-emergent laboratory abnormalities identified in the MURANO trial.
Table 12. Common (≥10%) New or Worsening Laboratory Abnormalities Occurring at ≥5% (Any Grade) or ≥2% (Grade 3 or 4) Higher Incidence with VEN+R Compared with B+R
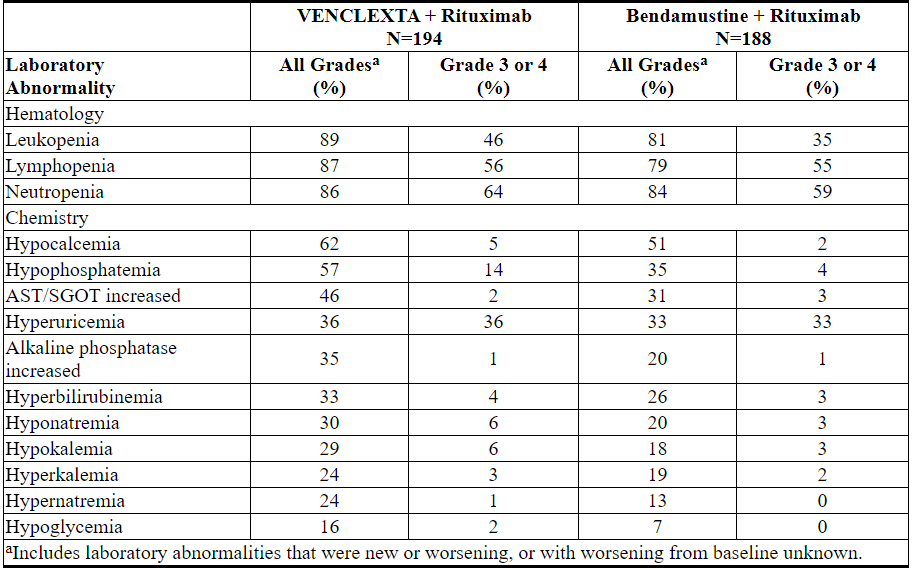
New Grade 4 laboratory abnormalities reported in ≥2% of patients treated with VEN+R included neutropenia (31%), lymphopenia (16%), leukopenia (6%), thrombocytopenia (6%), hyperuricemia (4%), hypocalcemia (2%), hypoglycemia (2%), and hypermagnesemia (2%).
Monotherapy Studies (M13-982, M14-032, and M12-175)
The safety of single agent VENCLEXTA at the 400 mg recommended daily dose following a dose ramp-up schedule is based on pooled data from three single-arm trials (M13-982, M14-032, and M12-175). In the pooled dataset, consisting of 352 patients with previously treated CLL or SLL, the median age was 66 years (range: 28 to 85 years), 93% were white, and 68% were male. The median number of prior therapies was 3 (range: 0 to 15). The median duration of treatment with VENCLEXTA at the time of data analysis was 14.5 months (range: 0 to 50 months). Fifty-two percent of patients received VENCLEXTA for more than 60 weeks.
Fatal adverse reactions that occurred in the absence of disease progression and within 30 days of venetoclax treatment were reported in 2% of patients in the VENCLEXTA monotherapy studies, most commonly (2 patients) from septic shock. Serious adverse reactions were reported in 52% of patients, with the most frequent (≥5%) being pneumonia (9%), febrile neutropenia (5%), and sepsis (5%).
Adverse reactions led to treatment discontinuation in 9% of patients, dose reduction in 13%, and dose interruption in 36%. The most frequent adverse reactions leading to drug discontinuation were thrombocytopenia and autoimmune hemolytic anemia. The most frequent adverse reaction (≥5%) leading to dose reductions or interruptions was neutropenia (8%).
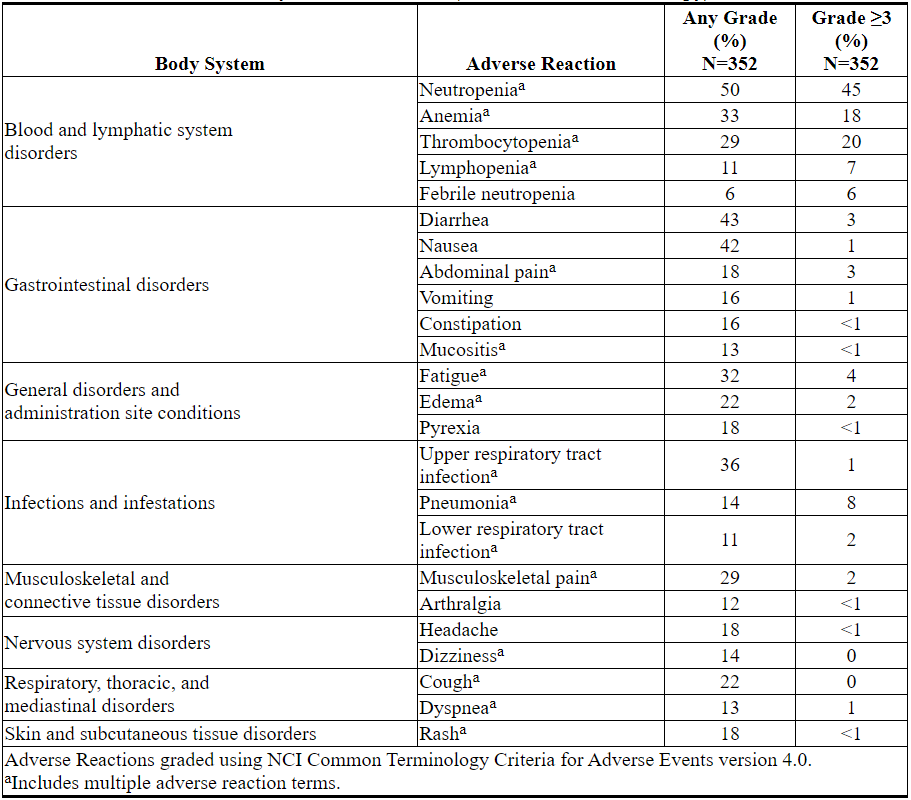
Adverse reactions identified in these trials of single-agent VENCLEXTA are presented in Table 13.
Table 13. Adverse Reactions Reported in ≥10% (Any Grade) or ≥5% (Grade ≥3) of Patients with Previously Treated CLL/SLL (VENCLEXTA Monotherapy)
Laboratory Abnormalities
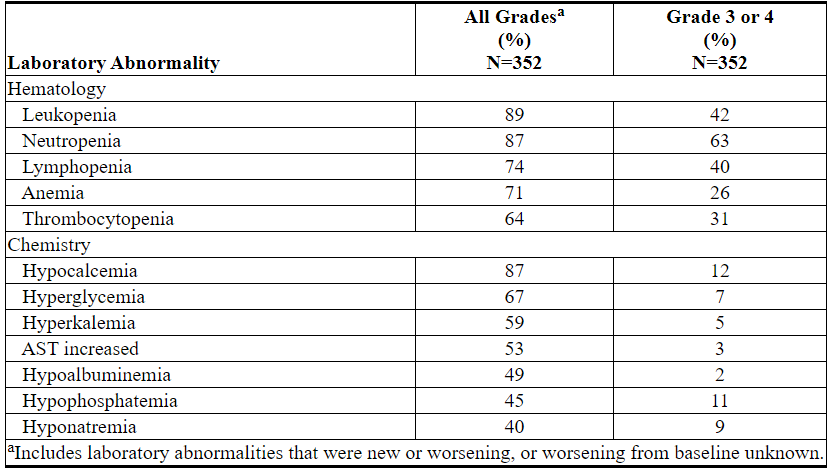
Table 14 describes common laboratory abnormalities reported throughout treatment that were new or worsening from baseline. The most common (>5%) Grade 4 laboratory abnormalities observed with VENCLEXTA monotherapy were hematologic laboratory abnormalities, including neutropenia (33%), leukopenia (11%), thrombocytopenia (15%), and lymphopenia (9%).
Table 14. New or Worsening Laboratory Abnormalities with VENCLEXTA Monotherapy (≥40% Any Grade or ≥10% Grade 3 or 4)
Important Adverse Reactions
Tumor Lysis Syndrome
Tumor lysis syndrome is an important identified risk when initiating VENCLEXTA.
CLL14
The incidence of TLS was 1% (3/212) in patients treated with VEN+G [see Warnings and Precautions (5.1)]. All three events of TLS resolved and did not lead to withdrawal from the study. Obinutuzumab administration was delayed in two cases in response to the TLS events.
MURANO
In the open-label randomized phase 3 study, the incidence of TLS was 3% (6/194) in patients treated with VEN+R. After 77/389 patients were enrolled in the study, the protocol was amended to incorporate the current TLS prophylaxis and monitoring measures described in sections 2.1 and 2.2 [see Dosage and Administration (2.1, 2.2)]. All events of TLS occurred during the VENCLEXTA ramp-up period and were resolved within two days. All six patients completed the ramp-up and reached the recommended daily dose of 400 mg of VENCLEXTA. No clinical TLS was observed in patients who followed the current 5-week ramp-up schedule and TLS prophylaxis and monitoring measures described in sections 2.1 and 2.2 [see Dosage and Administration (2.1, 2.2)]. Rates of laboratory abnormalities relevant to TLS for patients treated with VEN+R are presented in Table 12.
Monotherapy Studies (M13-982 and M14-032)
In 168 patients with CLL treated according to recommendations described in sections 2.1 and 2.2, the rate of TLS was 2% [see Dosage and Administration (2.1, 2.2)]. All events either met laboratory TLS criteria (laboratory abnormalities that met ≥2 of the following within 24 hours of each other: potassium >6 mmol/L, uric acid >476 µmol/L, calcium <1.75 mmol/L, or phosphorus >1.5 mmol/L); or were reported as TLS events. The events occurred in patients who had a lymph node(s) ≥5 cm and/or ALC ≥25 x 109/L. All events resolved within 5 days. No TLS with clinical consequences such as acute renal failure, cardiac arrhythmias or sudden death and/or seizures was observed in these patients. All patients had CLcr ≥50 mL/min. Laboratory abnormalities relevant to TLS were hyperkalemia (17% all Grades, 1% Grade ≥3), hyperphosphatemia (14% all Grades, 2% Grade ≥3), hypocalcemia (16% all Grades, 2% Grade ≥3), and hyperuricemia (10% all Grades, <1% Grade ≥3).
In the initial Phase 1 dose-finding trials, which had shorter (2-3 week) ramp-up phase and higher starting doses, the incidence of TLS was 13% (10/77; 5 laboratory TLS, 5 clinical TLS), including 2 fatal events and 3 events of acute renal failure, 1 requiring dialysis. After this experience, TLS risk assessment, dosing regimen, TLS prophylaxis and monitoring measures were revised [see Dosage and Administration (2.1, 2.2)].
6.2 Clinical Trial Experience with Acute Myeloid Leukemia
The safety of VENCLEXTA (400 mg daily dose) in combination with azacitidine (n=67) or decitabine (n= 13) and VENCLEXTA (600 mg daily dose) in combination with low-dose cytarabine (n= 61) is based on two non-randomized trials of patients with newly-diagnosed AML [see Clinical Studies (14.2)]. The median duration of exposure for patients taking VENCLEXTA in combination with azacitidine and decitabine was 6.5 months (range: 0.1 to 31.9 months) and 8.4 months (range: 0.5 to 22.3 months), respectively. The median duration of exposure for patients taking VENCLEXTA in combination with low dose cytarabine was 3.9 months (range: 0.2 to 29.2 months).
VENCLEXTA in Combination with Azacitidine or Decitabine
Azacitidine
The most common adverse reactions (≥30%) of any grade were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia.
Serious adverse reactions were reported in 75% of patients. The most frequent serious adverse reactions (≥5%) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome.
The incidence of fatal adverse drug reactions was 1.5% within 30 days of starting treatment. No reaction had an incidence of ≥2%.
Discontinuations due to adverse reactions occurred in 21% of patients. The most frequent adverse reactions leading to drug discontinuation (≥2%) were febrile neutropenia and pneumonia (excluding fungal).
Dosage interruptions due to adverse reactions occurred in 61% of patients. The most frequent adverse reactions leading to dose interruption (≥5%) were neutropenia, febrile neutropenia, and pneumonia (excluding fungal).
Dosage reductions due to adverse reactions occurred in 12% of patients. The most frequent adverse reaction leading to dose reduction (≥5%) was neutropenia.
Decitabine
The most common adverse reactions (≥30%) of any grade were febrile neutropenia, constipation, fatigue, thrombocytopenia, abdominal pain, dizziness, hemorrhage, nausea, pneumonia (excluding fungal), sepsis (excluding fungal), cough, diarrhea, neutropenia, back pain, hypotension, myalgia, oropharyngeal pain, peripheral edema, pyrexia, and rash.
Serious adverse reactions were reported in 85% of patients. The most frequent serious adverse reactions (≥5%) were febrile neutropenia, sepsis (excluding fungal), pneumonia (excluding fungal), diarrhea, fatigue, cellulitis, and localized infection.
One (8%) fatal adverse drug reaction of bacteremia occurred within 30 days of starting treatment.
Discontinuations due to adverse reactions occurred in 38% of patients. The most frequent adverse reaction leading to drug discontinuation (≥5%) was pneumonia (excluding fungal).
Dosage interruptions due to adverse reactions occurred in 62% of patients. The most frequent adverse reactions leading to dose interruption (≥5%) were febrile neutropenia, neutropenia, and pneumonia (excluding fungal).
Dosage reductions due to adverse reactions occurred in 15% of patients. The most frequent adverse reaction leading to dose reduction (≥5%) was neutropenia.
Adverse reactions reported in patients with newly-diagnosed AML using VENCLEXTA in combination with azacitidine or decitabine are presented in Table 15.
Table 15. Adverse Reactions Reported in ≥30% (Any Grade) or ≥5% (Grade ≥3) of Patients with AML Treated with VENCLEXTA in Combination with Azacitidine or Decitabine
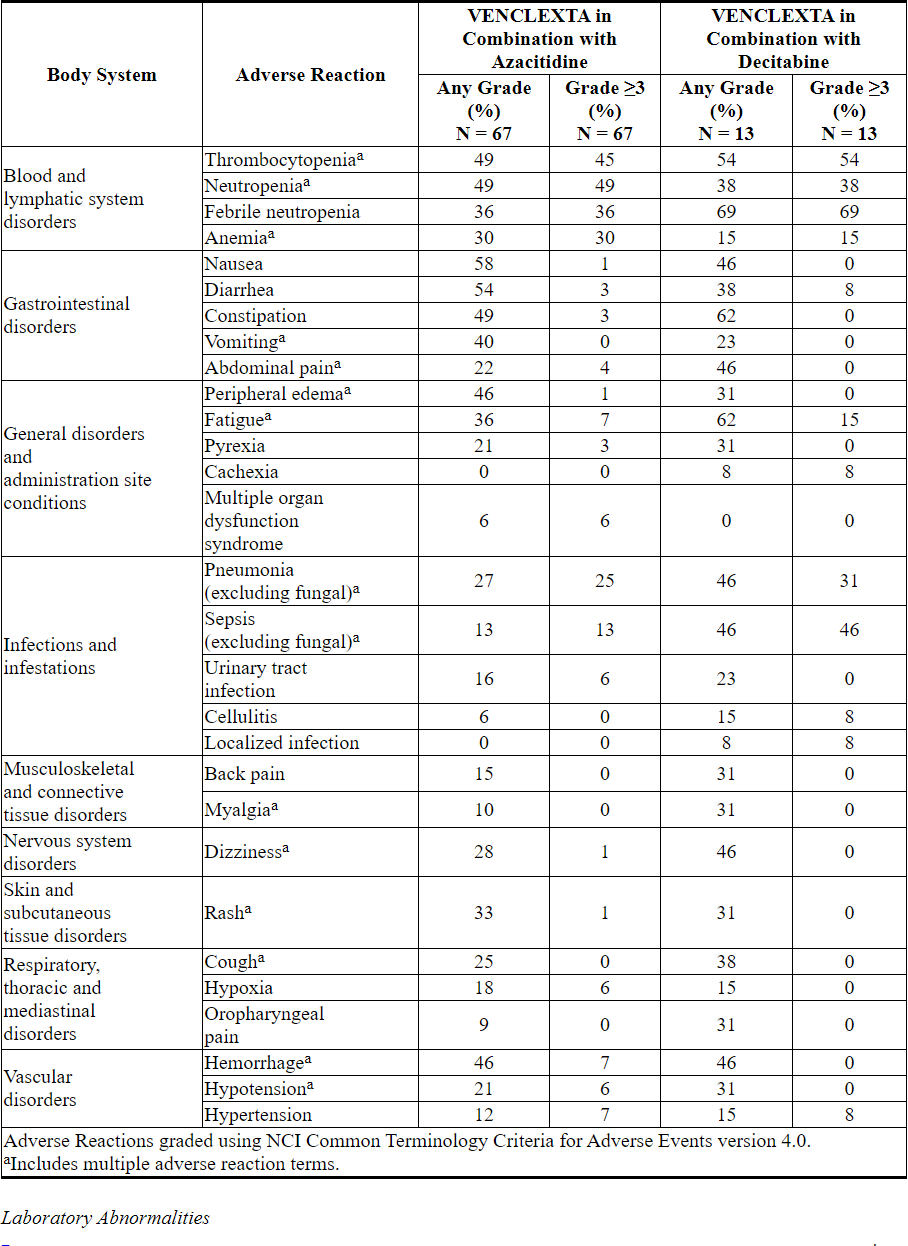
Table 16 describes common laboratory abnormalities reported throughout treatment that were new or worsening from baseline.
Table 16. New or Worsening Laboratory Abnormalities with VENCLEXTA Reported in ≥40% (Any Grade) or ≥10% (Grade 3 or 4) of Patients with AML Treated with VENCLEXTA in Combination with Azacitidine or Decitabine
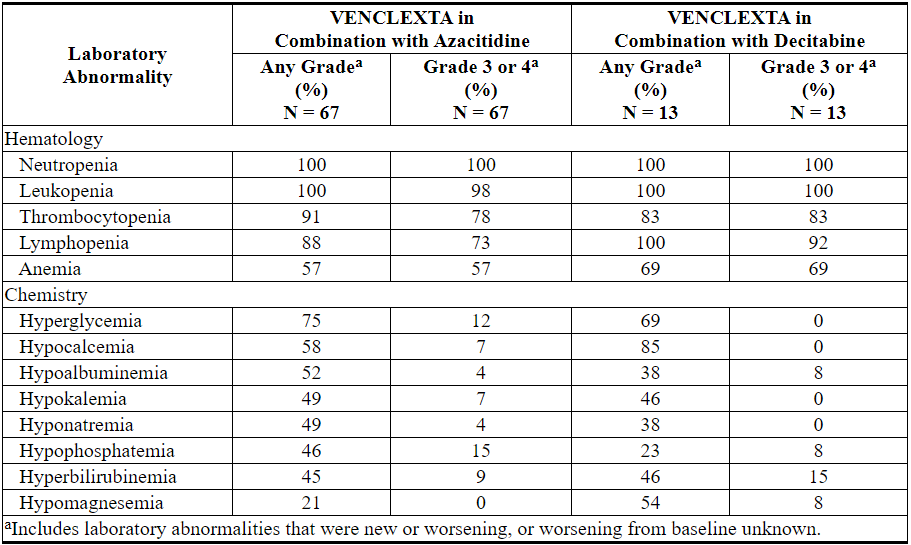
VENCLEXTA in Combination with Low-Dose Cytarabine
The most common adverse reactions (≥30%) of any grade were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea.
Serious adverse reactions were reported in 95% of patients. The most frequent serious adverse reactions (≥5%) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection.
The incidence of fatal adverse drug reactions was 4.9% within 30 days of starting treatment with no reaction having an incidence of ≥2%.
Discontinuations due to adverse reactions occurred in 33% of patients. The most frequent adverse reactions leading to drug discontinuation (≥2%) were hemorrhage and sepsis (excluding fungal).
Dosage interruptions due to adverse reactions occurred in 52% of patients. The most frequent adverse reactions leading to dose interruption (≥5%) were thrombocytopenia, neutropenia, and febrile neutropenia.
Dosage reductions due to adverse reactions occurred in 8% of patients. The most frequent adverse reaction leading to dose reduction (≥2%) was thrombocytopenia.
Adverse reactions reported in patients with newly-diagnosed AML receiving VENCLEXTA in combination with low-dose cytarabine are presented in Table 17.
Table 17. Adverse Reactions Reported in ≥30% (Any Grade) or ≥5% (Grade ≥3) of Patients with AML Treated with VENCLEXTA in Combination with Low-Dose Cytarabine
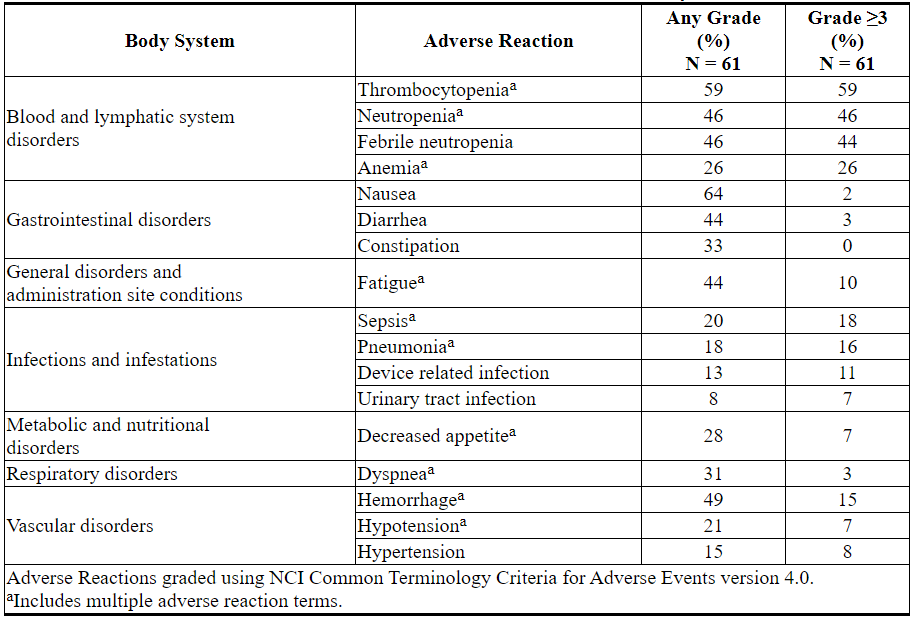
Laboratory Abnormalities
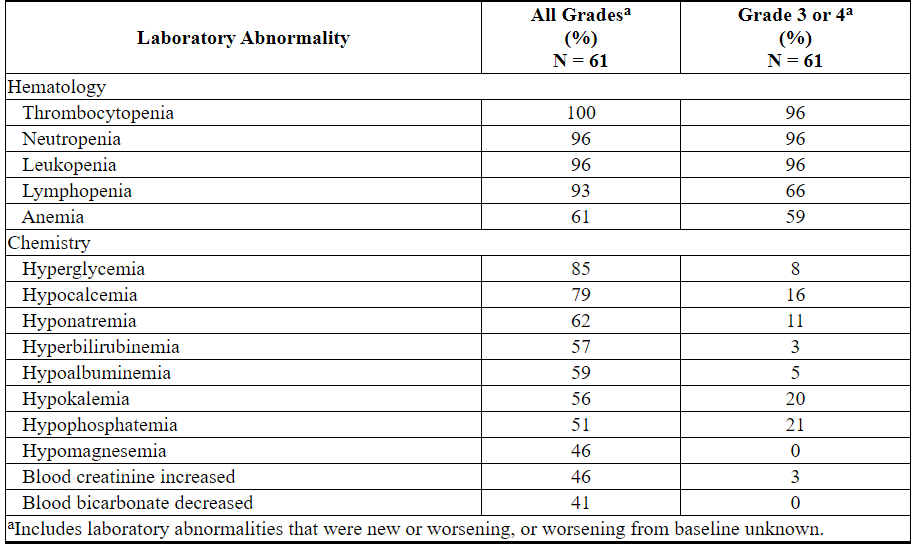
Table 18 describes common laboratory abnormalities reported throughout treatment that were new or worsening from baseline.
Table 18. New or Worsening Laboratory Abnormalities with VENCLEXTA Reported in ≥40% (Any Grade) or ≥10% (Grade 3 or 4) of Patients with AML Treated with VENCLEXTA in Combination with Low-Dose Cytarabine
Tumor Lysis Syndrome
Tumor lysis syndrome is an important risk when initiating treatment in patients with AML. The incidence of TLS was 3% (2/61) with VENCLEXTA in combination with low-dose cytarabine with implementation of dose ramp-up schedule in addition to standard prophylaxis and monitoring measures. All events were laboratory TLS, and all patients were able to reach the target dose.
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on VENCLEXTA
Strong or Moderate CYP3A Inhibitors or P-gp Inhibitors
Concomitant use with a strong or moderate CYP3A inhibitor or a P-gp inhibitor increases venetoclax Cmax and AUCinf[see Clinical Pharmacology (12.3)], which may increase VENCLEXTA toxicities, including the risk of TLS [see Warnings and Precautions (5)].
Concomitant use with a strong CYP3A inhibitor at initiation and during the ramp-up phase in patients with CLL/SLL is contraindicated [see Contraindications (4)].
In patients with CLL/SLL taking a steady daily dosage (after ramp-up phase), consider alternative medications or adjust VENCLEXTA dosage and closely monitor for signs of VENCLEXTA toxicities [see Dosage and Administration (2.3, 2.4)].
In patients with AML, adjust VENCLEXTA dosage and closely monitor for signs of VENCLEXTA toxicities [see Dosage and Administration (2.3, 2.4)].
Resume the VENCLEXTA dosage that was used prior to concomitant use with a strong or moderate CYP3A inhibitor or a P-gp inhibitor 2 to 3 days after discontinuation of the inhibitor [see Dosage and Administration (2.3, 2.4)].
Avoid grapefruit products, Seville oranges, and starfruit during treatment with VENCLEXTA, as they contain inhibitors of CYP3A.
Strong or Moderate CYP3A Inducers
Concomitant use with a strong CYP3A inducer decreases venetoclax Cmax and AUCinf[see Clinical Pharmacology (12.3)], which may decrease VENCLEXTA efficacy. Avoid concomitant use of VENCLEXTA with strong CYP3A inducers or moderate CYP3A inducers.
7.2 Effect of VENCLEXTA on Other Drugs
Warfarin
Concomitant use of VENCLEXTA increases warfarin Cmax and AUCinf[see Clinical Pharmacology (12.3)], which may increase the risk of bleeding. Closely monitor international normalized ratio (INR) in patients using warfarin concomitantly with VENCLEXTA.
P-gp Substrates
Concomitant use of VENCLEXTA increases Cmax and AUCinf of P-gp substrates [see Clinical Pharmacology (12.3)], which may increase toxicities of these substrates. Avoid concomitant use of VENCLEXTA with a P-gp substrate. If a concomitant use is unavoidable, separate dosing of the P-gp substrate at least 6 hours before VENCLEXTA.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on VENCLEXTA use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. Based on toxicity observed in mice, VENCLEXTA may cause fetal harm when administered to pregnant women. In mice, venetoclax was fetotoxic at exposures 1.2 times the human clinical exposure based on AUC at a human dose of 400 mg daily. If VENCLEXTA is used during pregnancy or if the patient becomes pregnant while taking VENCLEXTA, the patient should be apprised of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal data
In embryo-fetal development studies, venetoclax was administered to pregnant mice and rabbits during the period of organogenesis. In mice, venetoclax was associated with increased post-implantation loss and decreased fetal body weight at 150 mg/kg/day (maternal exposures approximately 1.2 times the human AUC exposure at a dose of 400 mg daily). No teratogenicity was observed in either the mouse or the rabbit.
8.2 Lactation
Risk Summary
There are no data on the presence of VENCLEXTA in human milk, the effects of VENCLEXTA on the breastfed child, or the effects of VENCLEXTA on milk production. Venetoclax was present in the milk when administered to lactating rats (see Data).
Because many drugs are excreted in human milk and because the potential for serious adverse reactions in a breastfed child from VENCLEXTA is unknown, advise nursing women to discontinue breastfeeding during treatment with VENCLEXTA.
Data
Animal Data
Venetoclax was administered (single dose; 150 mg/kg oral) to lactating rats 8 to 10 days parturition. Venetoclax in milk was 1.6 times lower than in plasma. Parent drug (venetoclax) represented the majority of the total drug-related material in milk, with trace levels of three metabolites.
8.3 Females and Males of Reproductive Potential
VENCLEXTA may cause fetal harm [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1)].
Pregnancy Testing
Conduct pregnancy testing in females of reproductive potential before initiation of VENCLEXTA [see Use in Specific Populations (8.1)].
Contraception
Advise females of reproductive potential to use effective contraception during treatment with VENCLEXTA and for at least 30 days after the last dose [see Use in Specific Populations (8.1)].
Infertility
Based on findings in animals, male fertility may be compromised by treatment with VENCLEXTA [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness have not been established in pediatric patients.
In a juvenile toxicology study, mice were administered venetoclax at 10, 30, or 100 mg/kg/day by oral gavage from 7 to 60 days of age. Clinical signs of toxicity included decreased activity, dehydration, skin pallor, and hunched posture at ≥30 mg/kg/day. In addition, mortality and body weight effects occurred at 100 mg/kg/day. Other venetoclax-related effects were reversible decreases in lymphocytes at ≥10 mg/kg/day; a dose of 10 mg/kg/day is approximately 0.06 times the clinical dose of 400 mg on a mg/m2 basis for a 20 kg child.
8.5 Geriatric Use
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Of the 352 patients with previously treated CLL/SLL evaluated for safety from 3 open-label trials of VENCLEXTA monotherapy, 57% (201/352) were ≥65 years of age and 18% (62/352) were ≥75 years of age.
No clinically meaningful differences in safety and effectiveness were observed between older and younger patients in the combination and monotherapy studies.
Acute Myeloid Leukemia
Of the 67 patients treated with VENCLEXTA in combination with azacitidine in the clinical trial, 96% were ≥65 years of age and 50% were ≥ 75 years of age. Of the 13 patients treated with VENCLEXTA in combination with decitabine in the clinical trial, 100% were ≥65 years of age and 26% were ≥ 75 years of age. Of the 61 patients treated with VENCLEXTA in combination with low-dose cytarabine, 97% were ≥65 years of age and 66% were ≥75 years of age.
The efficacy and safety data presented in the Adverse Reactions and Clinical Studies sections were obtained from these patients [see Adverse Reactions (6.2) and Clinical Studies (14.2)]. There are insufficient patient numbers to show differences in safety and effectiveness between geriatric and younger patients.
8.6 Renal Impairment
Due to the increased risk of TLS, patients with reduced renal function (CLcr <80 mL/min, calculated by Cockcroft-Gault formula) require more intensive prophylaxis and monitoring to reduce the risk of TLS when initiating treatment with VENCLEXTA [see Dosage and Administration (2.2, 2.3) and Warnings and Precautions (5.1)].
No dose adjustment is recommended for patients with mild or moderate renal impairment (CLcr ≥ 30 mL/min [see Clinical Pharmacology (12.3)]. A recommended dose has not been determined for patients with severe renal impairment (CLcr < 30 mL/min) or patients on dialysis.
8.7 Hepatic Impairment
No dose adjustment is recommended for patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment.
Reduce the dose of VENCLEXTA for patients with severe hepatic impairment (Child-Pugh C); monitor these patients more closely for signs of toxicity [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no specific antidote for VENCLEXTA. For patients who experience overdose, closely monitor and provide appropriate supportive treatment; during ramp-up phase interrupt VENCLEXTA and monitor carefully for signs and symptoms of TLS along with other toxicities [see Dosage and Administration (2.2, 2.3)]. Based on venetoclax large volume of distribution and extensive protein binding, dialysis is unlikely to result in significant removal of venetoclax.
11 DESCRIPTION
Venetoclax is a selective inhibitor of BCL-2 protein. It is a light yellow to dark yellow solid with the empirical formula C45H50ClN7O7S and a molecular weight of 868.44. Venetoclax has very low aqueous solubility. Venetoclax is described chemically as 4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide) and has the following chemical structure:
VENCLEXTA tablets for oral administration are supplied as pale yellow or beige tablets that contain 10, 50, or 100 mg venetoclax as the active ingredient. Each tablet also contains the following inactive ingredients: copovidone, colloidal silicon dioxide, polysorbate 80, sodium stearyl fumarate, and calcium phosphate dibasic. In addition, the 10 mg and 100 mg coated tablets include the following: iron oxide yellow, polyvinyl alcohol, polyethylene glycol, talc, and titanium dioxide. The 50 mg coated tablets also include the following: iron oxide yellow, iron oxide red, iron oxide black, polyvinyl alcohol, talc, polyethylene glycol and titanium dioxide. Each tablet is debossed with “V” on one side and “10”, “50” or “100” corresponding to the tablet strength on the other side.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Venetoclax is a selective and orally bioavailable small-molecule inhibitor of BCL-2, an anti-apoptotic protein. Overexpression of BCL-2 has been demonstrated in CLL and AML cells where it mediates tumor cell survival and has been associated with resistance to chemotherapeutics. Venetoclax helps restore the process of apoptosis by binding directly to the BCL-2 protein, displacing pro-apoptotic proteins like BIM, triggering mitochondrial outer membrane permeabilization and the activation of caspases. In nonclinical studies, venetoclax has demonstrated cytotoxic activity in tumor cells that overexpress BCL-2.
12.2 Pharmacodynamics
Based on the exposure response analyses for efficacy, a relationship between drug exposure and a greater likelihood of response was observed in clinical studies in patients with CLL/SLL, and in patients with AML. Based on the exposure response analyses for safety, a relationship between drug exposure and a greater likelihood of some safety events was observed in clinical studies in patients with AML. No exposure-safety relationship was observed in patients with CLL/SLL at doses up to 1200 mg given as monotherapy and up to 600 mg given in combination with rituximab.
Cardiac Electrophysiology
The effect of multiple doses of VENCLEXTA up to 1200 mg once daily (2 times the maximum approved recommended dosage) on the QTc interval was evaluated in an open-label, single-arm study in 176 patients with previously treated hematologic malignancies. VENCLEXTA had no large effect on QTc interval (i.e., > 20 ms) and there was no relationship between venetoclax exposure and change in QTc interval.
12.3 Pharmacokinetics
Venetoclax mean (± standard deviation) steady state Cmax was 2.1 ± 1.1 mcg/mL and AUC0-24 was32.8 ± 16.9 mcg•h/mL following administration of 400 mg once daily with a low-fat meal. Venetoclax steady state AUC increased proportionally over the dose range of 150 to 800 mg (0.25 to 1.33 times the maximum approved recommended dosage). The pharmacokinetics of venetoclax does not change over time.
Absorption
Maximum plasma concentration of venetoclax was reached 5 to 8 hours following multiple oral administration under fed conditions.
Effect of Food
Administration with a low-fat meal (approximately 512 kilocalories, 25% fat calories, 60% carbohydrate calories, and 15% protein calories) increased venetoclax exposure by approximately 3.4-fold and administration with a high-fat meal (approximately 753 kilocalories, 55% fat calories, 28% carbohydrate calories, and 17% protein calories) increased venetoclax exposure by 5.1- to 5.3-fold compared with fasting conditions.
Distribution
Venetoclax is highly bound to human plasma protein with unbound fraction in plasma <0.01 across a concentration range of 1-30 micromolar (0.87-26 mcg/mL). The mean blood-to-plasma ratio was 0.57. The apparent volume of distribution (Vdss/F) of venetoclax ranged from 256-321 L in patients.
Elimination
The terminal elimination half-life of venetoclax was approximately 26 hours.
Metabolism
Venetoclax is predominantly metabolized by CYP3A in vitro. The major metabolite identified in plasma, M27, has an inhibitory activity against BCL-2 that is at least 58-fold lower than venetoclax in vitro and its AUC represented 80% of the parent AUC.
Excretion
After single oral dose of radiolabeled [14C]-venetoclax 200 mg to healthy subjects, > 99.9% of the dose was recovered in feces (20.8% as unchanged) and < 0.1% in urine within 9 days.
Specific Populations
No clinically significant differences in the pharmacokinetics of venetoclax were observed based on age (19 to 90 years), sex, race (White, Black, Asians, and Others), weight, mild to moderate renal impairment (CLcr 30 to 89 mL/min, calculated by Cockcroft-Gault), or mild to moderate hepatic impairment (normal total bilirubin and aspartate transaminase (AST) > upper limit of normal (ULN) or total bilirubin 1 to 3 times ULN). The effect of severe renal impairment (CLcr < 30 mL/min) or dialysis on venetoclax pharmacokinetics is unknown.
Patients with Hepatic Impairment
Following a single dose of VENCLEXTA 50 mg, venetoclax systemic exposure (AUCinf) was 2.7-fold higher in subjects with severe hepatic impairment (Child-Pugh C) compared to subjects with normal hepatic function [see Dosage and Administration (2.5) and Use in Specific Populations (8.7)]. No clinically relevant differences in venetoclax systemic exposure were observed between subjects with mild or moderate hepatic impairment and subjects with normal hepatic function.
Drug Interactions Studies
Clinical Studies
No clinically significant differences in venetoclax pharmacokinetics were observed when co-administered with azacitidine, azithromycin, cytarabine, decitabine, gastric acid reducing agents, obinutuzumab, or rituximab.
Ketoconazole
Concomitant use of ketoconazole (a strong CYP3A, P-gp and BCRP inhibitor) 400 mg once daily for 7 days increased venetoclax Cmax by 130% and AUCinf by 540% [see Drug Interactions (7.1)].
Ritonavir
Concomitant use of ritonavir (a strong CYP3A, P-gp and OATP1B1/B3 inhibitor) 50 mg once daily for 14 days increased venetoclax Cmax by 140% and AUC by 690% [see Drug Interactions (7.1)].
Posaconazole
Concomitant use of posaconazole (a strong CYP3A and P-gp inhibitor) 300 mg with venetoclax 50 mg and 100 mg for 7 days resulted in 61% and 86% higher venetoclax Cmax, respectively, compared with venetoclax 400 mg administered alone. The venetoclax AUC24 was 90% and 144% higher, respectively.
Rifampin
Concomitant use of a single dose of rifampin(an OATP1B1/1B3 and P-gp inhibitor) 600 mg increased venetoclax Cmax by 106% and AUCinf by 78%. Concomitant use of multiple doses of rifampin (as a strong CYP3A inducer) 600 mg once daily for 13 days decreased venetoclax Cmax by 42% and AUCinf by 71% [see Drug Interactions (7.1)].
Warfarin
Concomitant use of a single 400 mg dose of venetoclax with 5 mg warfarin resulted in 18% to 28% increase in Cmax and AUCinf of R-warfarin and S-warfarin [see Drug Interactions (7.2)].
Digoxin
Concomitant use of a single dose of venetoclax 100 mg with digoxin (a P-gp substrate) 0.5 mg increased digoxin Cmax by 35% and AUCinf by 9% [see Drug Interactions (7.2)].
In Vitro Studies
Venetoclax is not an inhibitor or inducer of CYP1A2, CYP2B6, CYP2C19, CYP2D6 or CYP3A4. Venetoclax is a weak inhibitor of CYP2C8, CYP2C9, and UGT1A1.
Venetoclax is not an inhibitor of UGT1A4, UGT1A6, UGT1A9, or UGT2B7.
Venetoclax is an inhibitor and substrate of P-gp and BCRP and weak inhibitor of OATP1B1.
Venetoclax is not an inhibitor of OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, or MATE2K.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with venetoclax.
Venetoclax was not mutagenic in an in vitro bacterial mutagenicity (Ames) assay, did not induce numerical or structural aberrations in an in vitro chromosome aberration assay using human peripheral blood lymphocytes, and was not clastogenic in an in vivo mouse bone marrow micronucleus assay at doses up to 835 mg/kg. The M27 metabolite was negative for genotoxic activity in in vitro Ames and chromosome aberration assays.
Fertility and early embryonic development studies were conducted in male and female mice. These studies evaluate mating, fertilization, and embryonic development through implantation. There were no effects of venetoclax on estrous cycles, mating, fertility, corpora lutea, uterine implants or live embryos per litter at dosages up to 600 mg/kg/day. However, a risk to human male fertility exists based on testicular toxicity (germ cell loss) observed in dogs at exposures as low as 0.5 times the human AUC exposure at a dose of 400 mg.
13.2 Animal Toxicology and/or Pharmacology
In dogs, venetoclax caused single-cell necrosis in various tissues, including the gallbladder, exocrine pancreas, and stomach with no evidence of disruption of tissue integrity or organ dysfunction; these findings were minimal to mild in magnitude. Following a 4-week dosing period and subsequent 4-week recovery period, minimal single-cell necrosis was still present in some tissues and reversibility has not been assessed following longer periods of dosing or recovery.
In addition, after approximately 3 months of daily dosing in dogs, venetoclax caused progressive white discoloration of the hair coat, due to loss of melanin pigment.
14 CLINICAL STUDIES
14.1 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Combination Therapy
CLL14
CLL14 (BO25323) was a randomized (1:1), multicenter, open label, actively controlled, phase 3 trial (NCT02242942) that evaluated the efficacy and safety of VENCLEXTA in combination with obinutuzumab (VEN+G) versus obinutuzumab in combination with chlorambucil (GClb) for patients with previously untreated CLL with coexisting medical conditions (total Cumulative Illness Rating Scale [CIRS] score > 6 or CLcr < 70 mL/min). The trial required hepatic transaminases and total bilirubin ≤2 times upper limit of normal and excluded patients with Richter’s transformation or any individual organ/system impairment score of 4 by CIRS except eye, ear, nose, and throat organ system.
All patients received obinutuzumab at 1000 mg on Day 1 (the first dose could be split as 100 mg and 900 mg on Days 1 and 2), and on Days 8 and 15 of Cycle 1, and on Day 1 of each subsequent cycle, for a total of 6 cycles. Patients in the VEN+G arm began the 5-week VENCLEXTA ramp-up schedule [see Dosage and Administration (2.1, 2.2)] on Day 22 of Cycle 1, and received VENCLEXTA 400 mg once daily from Cycle 3 Day 1 until the last day of Cycle 12. Patients randomized to the GClb arm received 0.5 mg/kg oral chlorambucil on Day 1 and Day 15 of Cycles 1 to 12. Each cycle was 28 days.
A total of 432 patients were randomized, 216 to each study arm. Baseline demographic and disease characteristics were similar between the study arms. The median age was 72 years (range: 41 to 89 years), 89% were white, 67% were male; 36% and 43% were Binet stage B and C, respectively, and 88% had Eastern Cooperative Oncology Group (ECOG) performance status <2. The median CIRS score was 8.0 (range: 0 to 28) and 58% of patients had CLcr <70 mL/min. A 17p deletion was detected in 8% of patients, TP53 mutations in 7%, 11q deletion in 19%, and unmutated IgVH in 57%.
The major efficacy outcome was progression-free survival (PFS) as assessed by an Independent Review Committee (IRC). The median duration of follow-up for PFS was 28 months (range: 0.1 to 36 months).
Efficacy results for CLL14 are shown in Table 19. The Kaplan-Meier curve for PFS is shown in Figure 1.
Table 19. Efficacy Results in CLL14
Figure 1. Kaplan-Meier Curve of IRC-Assessed Progression-free Survival in CLL14
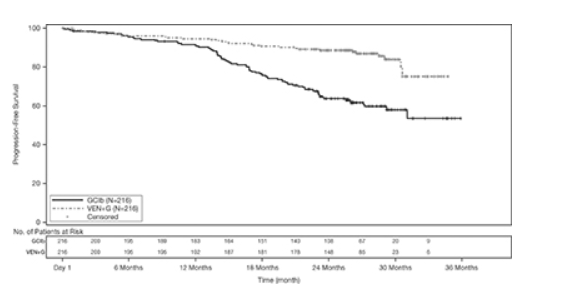
At the time of analysis, median overall survival (OS) had not been reached, with fewer than 10% of patients experiencing an event. The median duration of follow-up for OS was 28 months.
Minimal residual disease (MRD) was evaluated using allele-specific oligonucleotide polymerase chain reaction (ASO-PCR). The definition of negative status was less than one CLL cell per 104 leukocytes. Rates of MRD negativity 3 months after the completion of treatment regardless of response and in patients who achieved CR are shown in Table 20. At this assessment, 134 patients in the VEN+G arm who were MRD negative in peripheral blood had matched bone marrow specimens; of these, 122 patients (91%) were MRD negative in both peripheral blood and bone marrow.
Table 20. Minimal Residual Disease Negativity Rates Three Months After the Completion of Treatment in CLL14
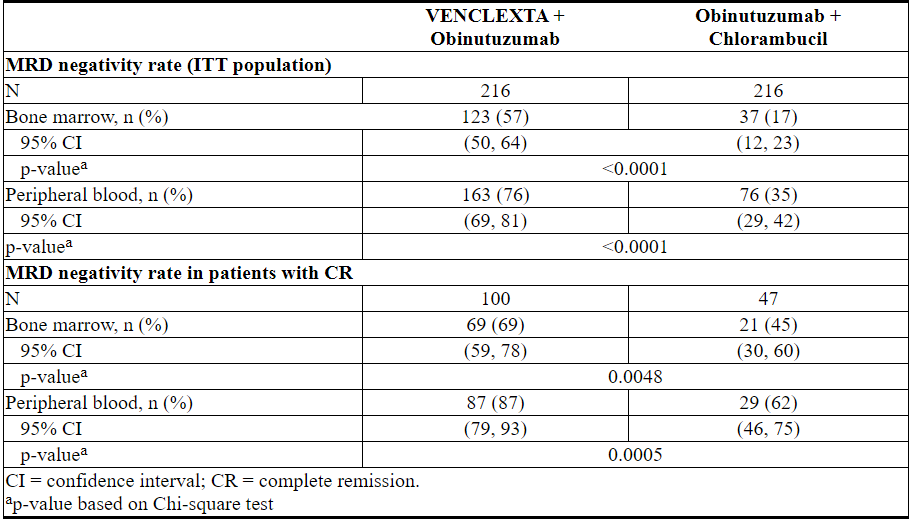
Twelve months after the completion of treatment, MRD negativity rates in peripheral blood were 58% (126/216) in patients treated with VEN+G and 9% (20/216) in patients treated with GClb.
MURANO
MURANO was a randomized (1:1), multicenter, open label study (NCT02005471) that evaluated the efficacy and safety of VENCLEXTA in combination with rituximab (VEN+R) versus bendamustine in combination with rituximab (B+R) in patients with CLL who had received at least one line of prior therapy. Patients in the VEN+R arm completed the 5-week ramp-up schedule [see Dosage and Administration (2.1, 2.2)] and received VENCLEXTA 400 mg once daily for 24 months from Cycle 1 Day 1 of rituximab in the absence of disease progression or unacceptable toxicity. Rituximab was initiated intravenously after the 5-week dose ramp-up at 375 mg/m2 on Day 1 of Cycle 1 and 500 mg/m2 on Day 1 of Cycles 2-6. Each cycle was 28 days. Patients randomized to B+R received bendamustine at 70 mg/m2 on Days 1 and 2 for 6 cycles (28-day cycle) and rituximab at the above described dose and schedule.
A total of 389 patients were randomized: 194 to the VEN+R arm and 195 to the B+R arm. Baseline demographic and disease characteristics were similar between the VEN+R and B+R arms. The median age was 65 years (range: 22 to 85 years), 97% were white, 74% were male, and 99% had ECOG performance status <2. Median prior lines of therapy was 1 (range: 1 to 5); 59% had received 1 prior therapy, 26% had received 2 prior therapies, and 16% had received 3 or more prior therapies. Prior therapies included alkylating agents (94%), anti-CD20 antibodies (77%), B-cell receptor pathway inhibitors (2%), and prior purine analogs (81%, including fludarabine/cyclophosphamide/rituximab in 55%). A 17p deletion was detected in 24% of patients, TP53 mutations in 25%, 11q deletion in 32%, and unmutated IgVH in 63%.
Efficacy was based on PFS as assessed by an IRC. The median follow-up for PFS was 23.4 months (range: 0 to 37.4+ months).
Efficacy results for MURANO are shown in Table 21. The Kaplan-Meier curve for PFS is shown in Figure 2.
Table 21. IRC-Assessed Efficacy Results in MURANO
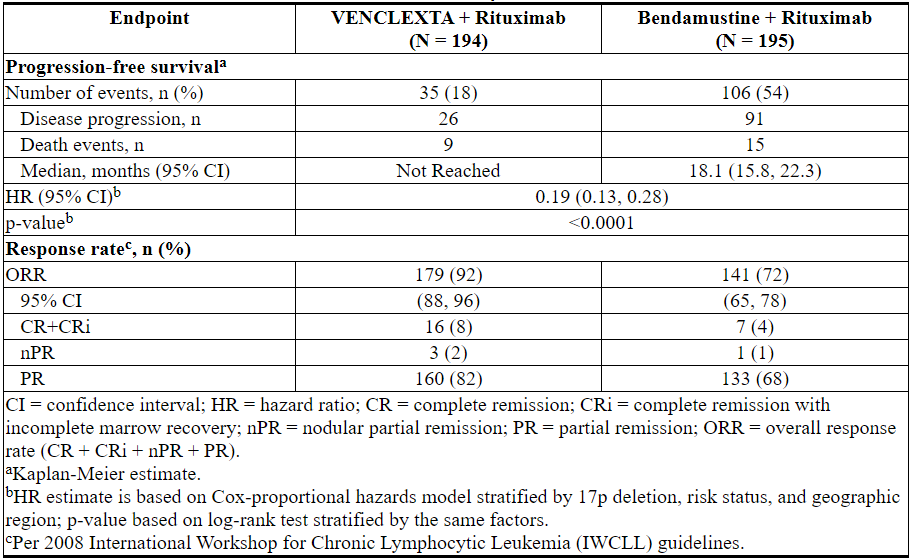
Figure 2. Kaplan-Meier Curve of IRC-Assessed Progression-free Survival in MURANO
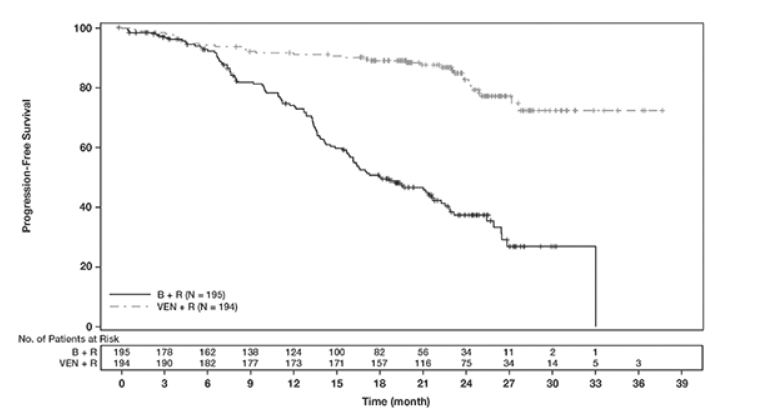
At the time of analysis, median overall survival had not been reached in either arm after a median follow-up of 22.9 months.
At 3 months after the last dose of rituximab, the MRD negativity rate in peripheral blood in patients who achieved PR or better was 53% (103/194) in the VEN+R arm and 12% (23/195) in the B+R arm. The MRD-negative CR/CRi rate at this timepoint was 3% (6/194) in the VEN+R arm and 2% (3/195) in the B+R arm.
Monotherapy
The efficacy of VENCLEXTA monotherapy in previously-treated CLL or SLL is based on three single-arm studies.
Study M13-982
The efficacy of VENCLEXTA was established in study M13-982 (NCT01889186), an open-label, single-arm, multicenter clinical trial of 106 patients with CLL with 17p deletion who had received at least one prior therapy. In the study, 17p deletion was confirmed in peripheral blood specimens from patients using Vysis CLL FISH Probe Kit, which is FDA approved for selection of patients for VENCLEXTA treatment. Patients received VENCLEXTA via a weekly ramp-up schedule starting at 20 mg and ramping to 50 mg, 100 mg, 200 mg and finally 400 mg once daily. Patients continued to receive 400 mg of VENCLEXTA orally once daily until disease progression or unacceptable toxicity.
Efficacy was based on overall response rate (ORR) as assessed by an IRC.
Table 22 summarizes the baseline demographic and disease characteristics of the study population.
Table 22. Baseline Patient Characteristics in Study M13-982
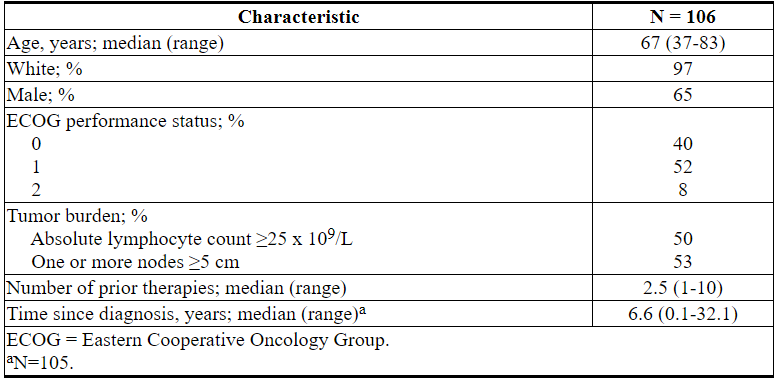
The median time on treatment at the time of evaluation was 12.1 months (range: 0 to 21.5 months). Efficacy results are shown in Table 23.
Table 23. Efficacy Results per IRC for Patients with Previously Treated CLL with 17p Deletion in Study M13-982
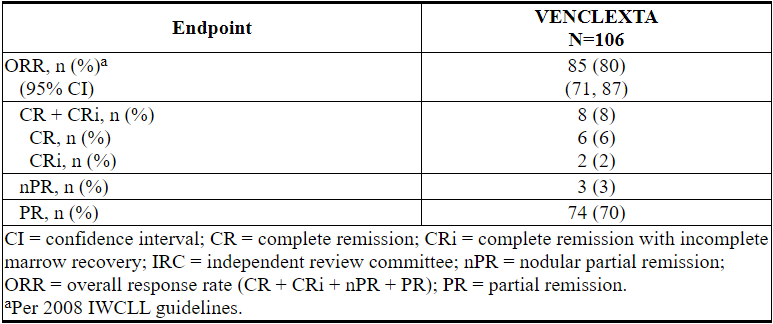
The median time to first response was 0.8 months (range: 0.1 to 8.1 months).
Based on a later data cutoff date and investigator-assessed efficacy, the duration of response (DOR) ranged from 2.9 to 32.8+ months. The median DOR has not been reached with median follow-up of 22 months.
Minimal residual disease was evaluated in peripheral blood and bone marrow for patients who achieved CR or CRi, following treatment with VENCLEXTA. Three percent (3/106) achieved MRD negativity in the peripheral blood and bone marrow (less than one CLL cell per 104 leukocytes).
Study M12-175
Study M12-175 (NCT01328626) was a multicenter, open-label trial that enrolled previously treated patients with CLL or SLL, including those with 17p deletion. Efficacy was evaluated in 67 patients (59 with CLL, 8 with SLL) who had received a 400 mg daily dose of VENCLEXTA. Patients continued this dose until disease progression or unacceptable toxicity. The median duration of treatment at the time of evaluation was 22.1 months (range: 0.5 to 71.7 months).
The median age was 65 years (range: 42 to 84 years), 78% were male and 87% were white. The median number of prior treatments was 3 (range: 1 to 11). At baseline, 67% of patients had one or more nodes ≥5 cm, 30% of patients had ALC ≥25 x 109/L, 33% had documented unmutated IgVH, and 21% had documented 17p deletion.
Efficacy was evaluated according to 2008 IWCLL guidelines and assessed by an IRC. The ORR was 76% (95% CI: 64%, 86%), CR + CRi rate was 10%, and PR rate was 66%. The median DOR was 36.2 months (range: 2.4 to 52.4 months).
Study M14-032
Study M14-032 (NCT02141282) was an open-label, multicenter, study that evaluated the efficacy of VENCLEXTA in patients with CLL who had been previously treated with and progressed on or after ibrutinib or idelalisib. Patients received a daily dose of 400 mg of VENCLEXTA following the ramp-up schedule. Patients continued to receive VENCLEXTA 400 mg once daily until disease progression or unacceptable toxicity. At the time of analysis, the median duration of treatment was 19.5 months (range: 0.1 to 39.5 months).
Of the 127 patients treated (91 with prior ibrutinib, 36 with prior idelalisib), the median age was 66 years (range: 28 to 85 years), 70% were male and 92% were white. The median number of prior treatments was 4 (range: 1 to 15). At baseline, 41% of patients had one or more nodes ≥5 cm, 31% had an absolute lymphocyte count ≥25 x 109/L, 57% had documented unmutated IgVH, and 39% had documented 17p deletion.
Efficacy was based on 2008 IWCLL guidelines and was assessed by an IRC. The ORR was 70% (95% CI: 61%, 78%), with a CR + CRi rate of 5%, and PR rate of 65%. The median DOR was not reached with a median follow-up time of 19.9 months (range: 2.9 to 36 months).
14.2 Acute Myeloid Leukemia
VENCLEXTA was studied in two open-label non-randomized trials in patients with newly-diagnosed AML who were ≥75 years of age, or had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline ECOG performance status of 2-3, severe cardiac or pulmonary comorbidity, moderate hepatic impairment, CLcr <45 mL/min, or other comorbidity. Efficacy was established based on the rate of complete remission (CR) and the duration of CR.
Study M14-358
VENCLEXTA was studied in a non-randomized, open-label clinical trial (NCT02203773) of VENCLEXTA in combination with azacitidine (N=84) or decitabine (N=31) in patients with newly-diagnosed AML. Of those patients, 67 who received azacitidine combination and 13 who received decitabine combination were age 75 years or older, or had comorbidities that precluded the use of intensive induction chemotherapy.
Patients received VENCLEXTA via a daily ramp-up to a final 400 mg once daily dose [see Dosage and Administration (2.1)]. During the ramp-up, patients received TLS prophylaxis and were hospitalized for monitoring. Azacitidine at 75 mg/m2 was administered either intravenously or subcutaneously on Days 1-7 of each 28-day cycle beginning on Cycle 1 Day 1. Decitabine at 20 mg/m2 was administered intravenously on Days 1-5 of each 28-day cycle beginning on Cycle 1 Day 1. Patients continued to receive treatment cycles until disease progression or unacceptable toxicity. Azacitidine dose reduction was implemented in the clinical trial for management of hematologic toxicity, see azacitidine full prescribing information. Dose reductions for decitabine were not implemented in the clinical trial.
Table 24 summarizes the baseline demographic and disease characteristics of the study population.
Table 24. Baseline Patient Characteristics for Patients with AML Treated with VENCLEXTA in Combination with Azacitidine or Decitabine
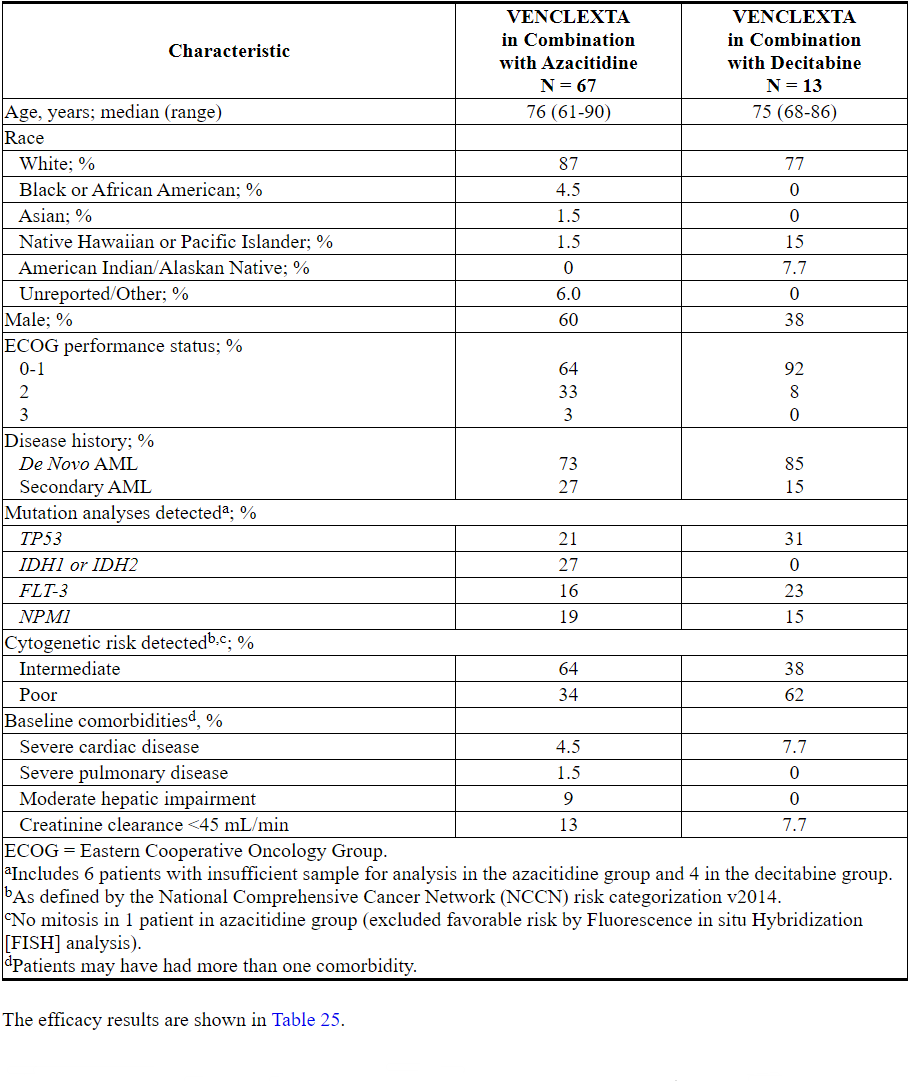
Table 25. Efficacy Results for Patients with Newly-Diagnosed AML Treated with VENCLEXTA in Combination with Azacitidine or Decitabine
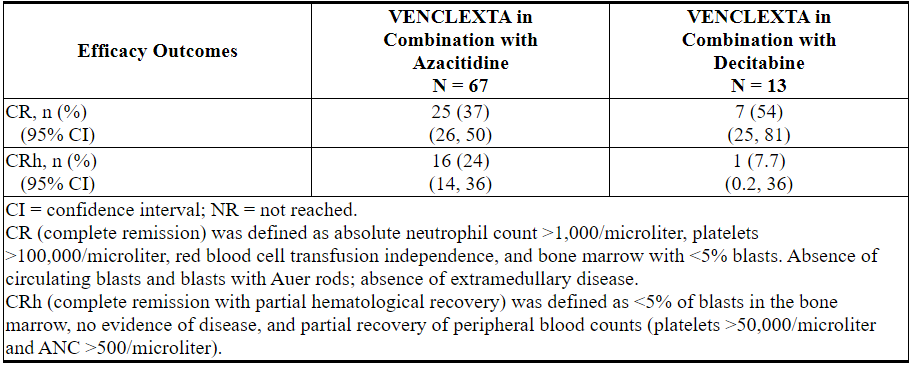
The median follow-up was 7.9 months (range: 0.4 to 36 months) for VENCLEXTA in combination with azacitidine. At the time of analysis, for patients who achieved a CR, the median observed time in remission was 5.5 months (range: 0.4 to 30 months). The observed time in remission is the time from the start of CR to the time of data cut-off date or relapse from CR.
The median follow-up was 11 months (range: 0.7 to 21 months) for VENCLEXTA in combination with decitabine. At the time of analysis, for patients who achieved a CR, the median observed time in remission was 4.7 months (range: 1.0 to 18 months). The observed time in remission is the time from the start of CR to the time of data cut-off date or relapse from CR.
Median time to first CR or CRh for patients treated with VENCLEXTA in combination with azacitidine was 1.0 month (range: 0.7 to 8.9 months).
Median time to first CR or CRh for patients treated with VENCLEXTA in combination with decitabine was 1.9 months (range: 0.8 to 4.2 months).
Of patients treated with VENCLEXTA in combination with azacitidine, 7.5% (5/67) subsequently received stem cell transplant.
The study enrolled 35 additional patients (age range: 65 to 74 years) who did not have known comorbidities that preclude the use of intensive induction chemotherapy and were treated with VENCLEXTA in combination with azacitidine (N=17) or decitabine (N=18).
For the 17 patients treated with VENCLEXTA in combination with azacitidine, the CR rate was 35% (95% CI: 14%, 62%). The CRh rate was 41% (95% CI: 18%, 67%). Seven (41%) patients subsequently received stem cell transplant.
For the 18 patients treated with VENCLEXTA in combination with decitabine, the CR rate was 56% (95% CI: 31%, 79%). The CRh rate was 22% (95% CI: 6.4%, 48%). Three (17%) patients subsequently received stem cell transplant.
Study M14-387
VENCLEXTA was studied in a non-randomized, open-label clinical trial (NCT02287233) of VENCLEXTA in combination with low dose cytarabine (N=82) in patients with newly-diagnosed AML, including patients with previous exposure to a hypomethylating agent for an antecedent hematologic disorder. Of those patients, 61 were age 75 years or older, or had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline ECOG performance status of 2-3, severe cardiac or pulmonary comorbidity, moderate hepatic impairment, CLcr ≥30 to <45 mL/min, or other comorbidity.
Patients initiated VENCLEXTA via daily ramp-up to a final 600 mg once daily dose [see Dosage and Administration (2.1)]. During the ramp-up, patients received TLS prophylaxis and were hospitalized for monitoring. Cytarabine at a dose of 20 mg/m2 was administered subcutaneously once daily on Days 1-10 of each 28-day cycle beginning on Cycle 1 Day 1. Patients continued to receive treatment cycles until disease progression or unacceptable toxicity. Dose reduction for low-dose cytarabine was not implemented in the clinical trial.
Table 26 summarizes the baseline demographic and disease characteristics of the study population.
Table 26. Baseline Patient Characteristics for Patients with AML Treated with VENCLEXTA in Combination with Low-Dose Cytarabine
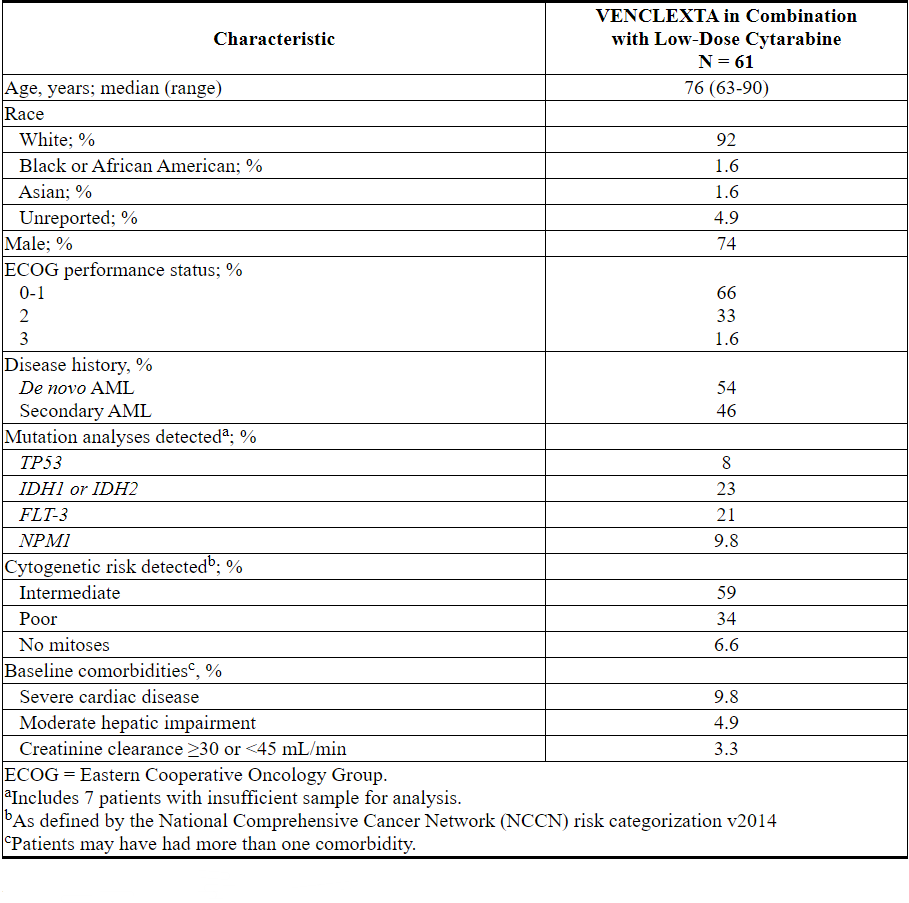
Efficacy results are shown in Table 27.
Table 27. Efficacy Results for Patients with Newly-Diagnosed AML Treated with VENCLEXTA in Combination with Low-Dose Cytarabine
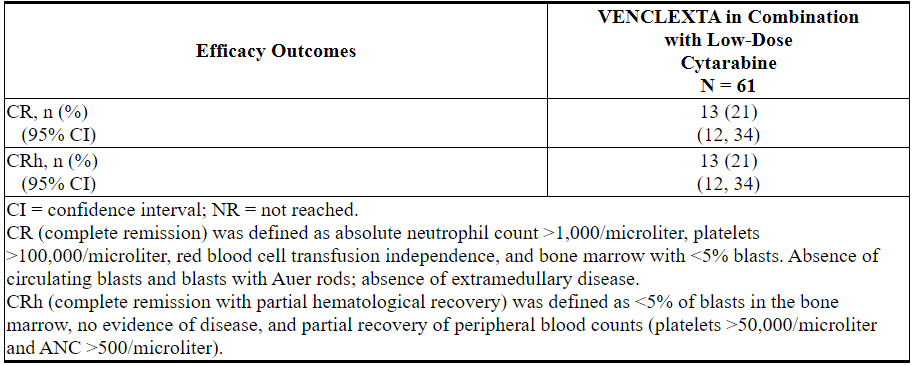
The median follow-up was 6.5 months (range: 0.3 to 34 months). At the time of analysis, for patients who achieved a CR, the median observed time in remission was 6.0 months (range: 0.03 to 25 months). The observed time in remission is the time from the start of CR to the time of data cut-off date or relapse from CR.
Median time to first CR or CRh for patients treated with VENCLEXTA in combination with low-dose cytarabine was 1.0 month (range: 0.8 to 9.4 months).
The study enrolled 21 additional patients (age range: 67 to 74 years) who did not have known comorbidities that preclude the use of intensive induction chemotherapy and were treated with VENCLEXTA in combination with low-dose cytarabine. The CR rate was 33% (95% CI: 15%, 57%). The CRh rate was 24% (95% CI: 8.2%, 47%). One patient (4.8%) subsequently received stem cell transplant.
16 HOW SUPPLIED/STORAGE AND HANDLING
VENCLEXTA is dispensed as follows:
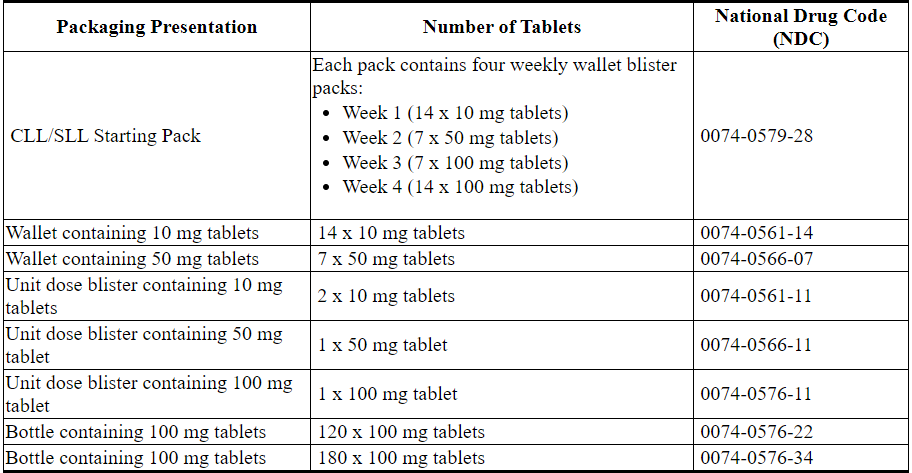
VENCLEXTA 10 mg film-coated tablets are round, biconvex shaped, pale yellow debossed with “V” on one side and “10” on the other side.
VENCLEXTA 50 mg film-coated tablets are oblong, biconvex shaped, beige debossed with “V” on one side and “50” on the other side.
VENCLEXTA 100 mg film-coated tablets are oblong, biconvex shaped, pale yellow debossed with “V” on one side and “100” on the other side.
Store at or below 86°F (30°C).
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
- Tumor Lysis Syndrome
Advise patients of the potential risk of TLS, particularly at treatment initiation and during ramp-up phase, and to immediately report any signs and symptoms associated with this event (fever, chills, nausea, vomiting, confusion, shortness of breath, seizure, irregular heartbeat, dark or cloudy urine, unusual tiredness, muscle pain, and/or joint discomfort) to their health care provider (HCP) for evaluation [see Warnings and Precautions (5.1)].
Advise patients to be adequately hydrated every day when taking VENCLEXTA to reduce the risk of TLS. The recommended volume is 6 to 8 glasses (approximately 56 ounces total) of water each day. Patients should drink water starting 2 days before and on the day of the first dose, and every time the dose is increased [see Dosage and Administration (2.2)].
Advise patients of the importance of keeping scheduled appointments for blood work or other laboratory tests [see Dosage and Administration (2.2)].
Advise patients that it may be necessary to take VENCLEXTA in the hospital or medical office setting to allow monitoring for TLS. - Neutropenia
Advise patients to contact their HCP immediately if they develop a fever or any signs of infection. Advise patients of the need for periodic monitoring of blood counts [see Warnings and Precautions (5.2)]. - Infections
Advise patients to contact their HCP immediately if they develop a fever or any signs of infection [see Warnings and Precautions (5.3)]. - Drug Interactions
Advise patients to avoid consuming grapefruit products, Seville oranges, or starfruit during treatment with VENCLEXTA. Advise patients that VENCLEXTA may interact with some drugs; therefore, advise patients to inform their health care provider of the use of any prescription medication, over-the-counter drugs, vitamins and herbal products [see Contraindications (4) and Drug Interactions (7.1)]. - Immunizations
Advise patients to avoid vaccination with live vaccines because they may not be safe or effective during treatment with VENCLEXTA [see Warnings and Precautions (5.4)]. - Pregnancy and Lactation
Advise women of the potential risk to the fetus and to avoid pregnancy during treatment with VENCLEXTA. Advise female patients of reproductive potential to use effective contraception during therapy and for at least 30 days after completing of therapy. Advise females to contact their HCP if they become pregnant, or if pregnancy is suspected, during treatment with VENCLEXTA. Also advise patients not to breastfeed while taking VENCLEXTA [see Warnings and Precautions (5.5), and Use in Specific Populations (8.1, 8.2, and 8.3)]. - Male Infertility
Advise patients of the possibility of infertility and possible use of sperm banking for males of reproductive potential [see Use in Specific Populations (8.3)].
Instructions for Taking VENCLEXTA
Advise patients to take VENCLEXTA exactly as prescribed and not to change their dose or to stop taking VENCLEXTA unless they are told to do so by their HCP. Advise patients to take VENCLEXTA orally once daily, at approximately the same time each day, according to their HCP's instructions and that the tablets should be swallowed whole with a meal and water without being chewed, crushed, or broken [see Dosage and Administration (2.1)].
Advise patients with CLL/SLL to keep VENCLEXTA in the original packaging during the first 4 weeks of treatment, and not to transfer the tablets to a different container.
Advise patients that if a dose of VENCLEXTA is missed by less than 8 hours, to take the missed dose right away and take the next dose as usual. If a dose of VENCLEXTA is missed by more than 8 hours, advise patients to wait and take the next dose at the usual time [see Dosage and Administration (2.6)].
Advise patients not to take any additional dose that day if they vomit after taking VENCLEXTA, and to take the next dose at the usual time the following day.
Manufactured and Marketed by:
AbbVie Inc.
North Chicago, IL 60064and
Marketed by:
Genentech USA, Inc.
A Member of the Roche Group
South San Francisco, CA 94080-4990© 2019 AbbVie Inc.
© 2019 Genentech, Inc.
03-B947 July 2019MEDICATION GUIDE
VENCLEXTA® (ven-KLEKS-tuh)
(venetoclax tablets)



CLL/SLL Starting Pack
VENCLEXTA®
(venetoclax tablets)
10 mg, 50 mg, and 100 mg
Starting Pack
! WARNING
Contact your doctor when you receive this medication.
It may be necessary to take your first dose in the presence of your doctor to prevent a potential serious side effect.
DISPENSER: Each time VENCLEXTA is dispensed give the patient the enclosed Medication Guide.
abbvie
Rx only
Genentech
NDC 0074–0561–14
Rx only
VENCLEXTA®
(venetoclax tablets)
10 mg
14 Tablets
Dispense the accompanying Medication Guide to each patient.
abbvie
Genentech

NDC 0074-0566-11
Rx only
VENCLEXTA®
(venetoclax tablets)
50 mg
1 Tablet
Dispense the accompanying Medication Guide to each patient.
abbvie
Genentech
NDC 0074-0576-22
Rx only
VENCLEXTA®
(venetoclax tablets)
100 mg
120 Tablets
Do not accept if seal over bottle opening is broken or missing.
Each film-coated tablet contains 100 mg of venetoclax.
Keep out of reach of children.
Store at or below 86°F (30°C).
abbvie
Genentech

NDC 0074-0576-22
Rx only
VENCLEXTA®
(venetoclax tablets)
100 mg
120 Tablets
Dispense the accompanying Medication Guide to each patient.
abbvie
Genetech

NDC 0074–0576–11
Rx only
VENCLEXTA®
(venetoclax tablets)
100 mg
1 Tablet
Dispense the accompanying Medication Guide to each patient.
abbvie
Genetech
【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。