商品详情
返回产品目录





商品包装及说明书因厂家更换频繁,如有不符以实物为主
索托雷塞片
国际零售参考价:¥**/盒
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。
1 适应症及用途
LUMAKRAS 适用于治疗患有KRAS G12C突变的局部晚期或转移性非小细胞肺癌 (NSCLC) 的成年患者,根据 FDA 批准的测试确定[见剂量和给药方法(2.1) ],这些患者已在至少接受过一次全身治疗。
该适应症根据总体缓解率(ORR)和缓解持续时间(DOR)在加速批准下获得批准[见临床研究(14) ]。该适应症的持续批准可能取决于验证性试验中临床益处的验证和描述。
2 剂量和给药方法
2.1 患者选择
根据肿瘤或血浆标本中KRAS G12C突变的存在,选择用 LUMAKRAS 治疗局部晚期或转移性 NSCLC 的患者[见临床研究(14) ]。如果血浆样本中未检测到突变,则测试肿瘤组织。
有关 FDA 批准的KRAS G12C突变检测测试的信息,请访问:http://www.fda.gov/CompanionDiagnostics。
2.2 推荐剂量和给药方法
LUMAKRAS 的推荐剂量为 960 mg(三片 320 mg 片剂或八片 120 mg 片剂),每天口服一次,直至疾病进展或出现不可接受的毒性。
每天在同一时间随食物或不随食物服用LUMAKRAS日剂量[见临床药理学(12.3) ]。整个吞下药片。不要咀嚼、压碎或劈开药片。如果错过一剂 LUMAKRAS 超过 6 小时,请在第二天按照处方服用下一剂。不要同时服用 2 剂以弥补错过的剂量。
如果服用 LUMAKRAS 后出现呕吐,请勿服用额外剂量。第二天按照规定服用下一剂。
吞咽固体困难的患者的给药
将片剂分散在 120 毫升(4 盎司)非碳酸室温水中,无需压碎。不应使用其他液体。搅拌或旋转杯子约 3 分钟,直至药片分散成小块(药片不会完全溶解),然后立即或在 2 小时内饮用。混合物的外观可以从浅黄色到亮黄色。吞下片剂分散液。不要咀嚼药片碎片。再用 120 毫升(4 盎司)水和饮料冲洗容器。如果混合物没有立即消耗,请再次搅拌混合物以确保药片分散。
2.3 不良反应的剂量调整
表 1 总结了 LUMAKRAS 剂量减少水平。表 2 提供了针对不良反应的剂量修改。
如果发生不良反应,最多允许减少两次剂量。如果患者无法耐受每日一次 240 mg 的最低剂量,请停用 LUMAKRAS。
表 1. 针对不良反应推荐的 LUMAKRAS 剂量减少水平
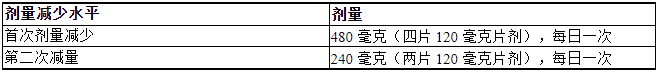
表 2. 针对不良反应推荐的 LUMAKRAS 剂量修改
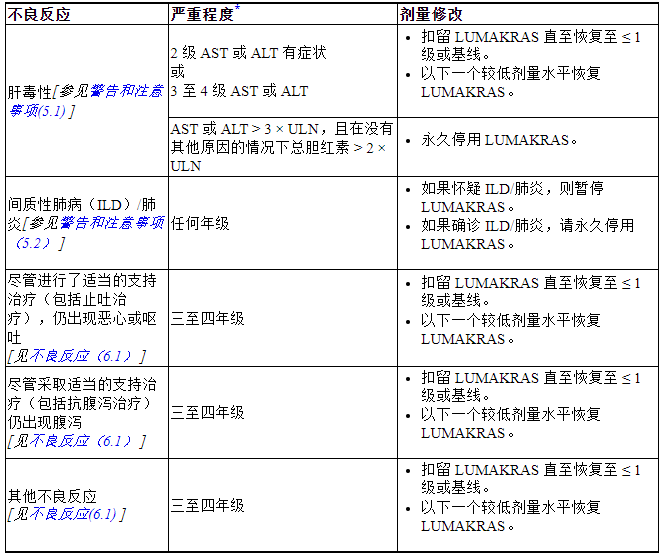
ALT = 丙氨酸转氨酶;AST = 天冬氨酸转氨酶;ULN = 正常上限
*根据美国国家癌症研究所不良事件通用术语标准 (NCI CTCAE) 5.0 版定义的分级
2.4 LUMAKRAS 与酸还原剂的共同给药
避免质子泵抑制剂 (PPI) 和 H 2受体拮抗剂与 LUMAKRAS 共同给药。如果无法避免用减酸剂治疗,在局部抗酸剂给药前4小时或给药后10小时服用LUMAKRAS [见药物相互作用(7.1)和临床药理学(12.3) ]。
3 剂型和规格
- 片剂:320 mg,米色,椭圆形,立即释放,薄膜包衣,一侧凹陷有“AMG”,另一侧凹陷有“320”。
- 片剂:120 mg,黄色,长方形,立即释放,薄膜包衣,一侧凹陷有“AMG”,另一侧凹陷有“120”。
4 禁忌症
没有任何。
5 警告和注意事项
5.1 肝毒性
LUMAKRAS 可引起肝毒性,可能导致药物性肝损伤和肝炎。在CodeBreaK 100中接受LUMAKRAS的357例患者中[见不良反应(6.1) ],肝毒性发生率为1.7%(所有等级)和1.4%(3级)。接受 LUMAKRAS 治疗的患者中,共有 18% 的丙氨酸转氨酶 (ALT)/天冬氨酸转氨酶 (AST) 升高;6% 为 3 级,0.6% 为 4 级。首次出现 ALT/AST 升高的中位时间为 9 周(范围:0.3 至 42)。7% 的患者发生 ALT/AST 升高导致剂量中断或减少的情况。由于 2.0% 的患者 ALT/AST 升高,LUMAKRAS 被停用。除了中断或减少剂量外,5%的患者还接受皮质类固醇治疗肝毒性。
在开始 LUMAKRAS 之前监测肝功能测试(ALT、AST 和总胆红素),治疗前 3 个月每 3 周监测一次,然后每月一次或根据临床指示,对出现转氨酶和/或胆红素升高。根据不良反应的严重程度暂停、减少剂量或永久终止LUMAKRAS [见剂量和给药方法(2.3)和不良反应(6.1) ]。
5.2 间质性肺病(ILD)/肺炎
LUMAKRAS 可引起致命的 ILD/肺炎。在 CodeBreaK 100 中接受 LUMAKRAS 的 357 例患者中[见不良反应(6.1) ],0.8%患者发生 ILD/肺炎,所有病例发病时为 3 或 4 级,1 例死亡。ILD/肺炎首次发病的中位时间为 2 周(范围:2 至 18 周)。0.6% 的患者因 ILD/肺炎而停用 LUMAKRAS。监测患者是否出现新的或恶化的 ILD/肺炎肺部症状(例如呼吸困难、咳嗽、发烧)。对疑似 ILD/肺炎患者立即停用 LUMAKRAS,如果没有发现 ILD/肺炎的其他潜在原因,则永久终止 LUMAKRAS [见剂量和给药方法(2.3)和不良反应(6.1) ]。
6 不良反应
标签的其他部分更详细地讨论了以下具有临床意义的不良反应:
- 肝毒性[参见警告和注意事项(5.1) ]
- 间质性肺病(ILD)/肺炎[参见警告和注意事项(5.2) ]
6.1 临床试验经验
由于临床试验是在广泛不同的条件下进行的,一种药物临床试验中观察到的不良反应率不能直接与另一种药物临床试验中的发生率进行比较,并且可能无法反映实践中观察到的发生率。
警告和注意事项中描述的汇总安全人群反映了CodeBreaK 100 中登记的357 名患有KRAS G12C突变的非小细胞肺癌和其他实体瘤患者暴露于 LUMAKRAS 作为单药 960 mg 口服,每日一次,其中 28% 暴露了 6 个月或更长,3% 的暴露时间超过一年。
非小细胞肺癌
在 CodeBreaK 100 中,在KRAS G12C突变局部晚期或转移性 NSCLC患者的子集中评估了 LUMAKRAS 的安全性[见临床研究(14) ]。患者每天口服一次 LUMAKRAS 960 mg,直至疾病进展或出现不可接受的毒性 (n = 204)。在接受 LUMAKRAS 治疗的患者中,39% 的患者暴露时间为 6 个月或更长时间,3% 的患者暴露时间超过一年。
接受 LUMAKRAS 治疗的患者的中位年龄为 66 岁(范围:37 至 86 岁);55% 女性;80% 白人、15% 亚洲人、3% 黑人。
50% 接受 LUMAKRAS 治疗的患者出现严重不良反应。≥2%患者的严重不良反应为肺炎(8%)、肝毒性(3.4%)和腹泻(2%)。接受 LUMAKRAS 治疗的患者中有 3.4% 发生致命不良反应,包括呼吸衰竭 (0.8%)、肺炎 (0.4%)、心脏骤停 (0.4%)、心力衰竭 (0.4%)、胃溃疡 (0.4%) 和肺炎(0.4%)。
9% 的患者因不良反应而永久停用 LUMAKRAS。导致 ≥ 2% 患者永久停用 LUMAKRAS 的不良反应包括肝毒性 (4.9%)。
34% 的患者因不良反应而中断 LUMAKRAS 剂量。≥2%的患者需要中断用药的不良反应为肝毒性(11%)、腹泻(8%)、肌肉骨骼疼痛(3.9%)、恶心(2.9%)和肺炎(2.5%)。
5% 的患者因不良反应而减少 LUMAKRAS 剂量。≥ 2% 的患者需要减少剂量的不良反应包括 ALT 升高 (2.9%) 和 AST 升高 (2.5%)。
最常见的不良反应(≥20%)是腹泻、肌肉骨骼疼痛、恶心、疲劳、肝毒性和咳嗽。最常见的实验室异常(≥25%)是淋巴细胞减少、血红蛋白减少、天冬氨酸转氨酶增加、丙氨酸转氨酶增加、钙减少、碱性磷酸酶增加、尿蛋白增加和钠减少。
表 3 总结了 CodeBreaK 100 中观察到的常见不良反应。
表 3. 在 CodeBreaK 100 中接受 LUMAKRAS 治疗的 KRAS G12C 突变 NSCLC 患者的不良反应 (≥ 10%) *
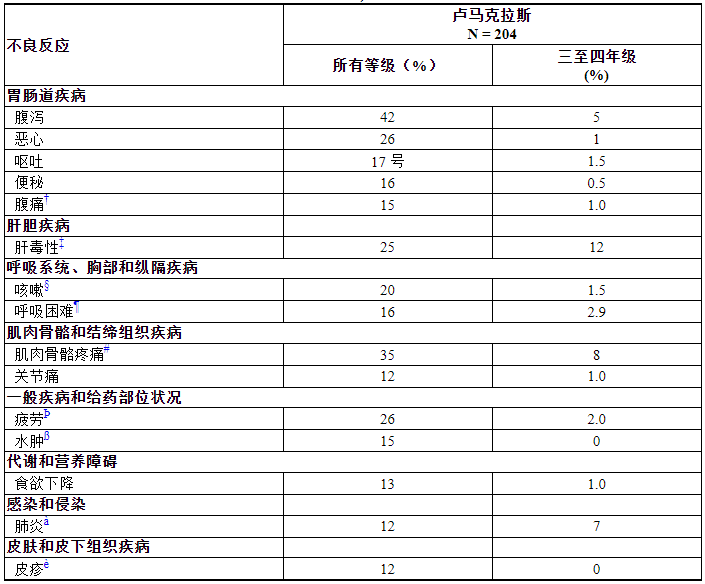
*由 NCI CTCAE 5.0 版定义的分级。
†腹痛包括腹痛、上腹痛、下腹痛。
‡肝毒性包括丙氨酸转氨酶升高、天冬氨酸转氨酶升高、血胆红素升高、药物性肝损伤、肝炎、肝毒性、肝功能检查升高、转氨酶升高等。
§咳嗽包括咳嗽、排痰性咳嗽和上呼吸道咳嗽综合征。
¶呼吸困难包括劳力性呼吸困难和劳力性呼吸困难。
#肌肉骨骼疼痛包括背痛、骨痛、肌肉骨骼胸痛、肌肉骨骼不适、肌肉骨骼疼痛、肌痛、颈部疼痛、非心源性胸痛和四肢疼痛。
Þ疲劳包括乏力和乏力。
β水肿包括全身性水肿、局部性水肿、周围性水肿、眶周水肿、睾丸水肿。
A肺炎包括肺炎、吸入性肺炎、细菌性肺炎和葡萄球菌性肺炎。
è皮疹包括皮炎、痤疮样皮炎、皮疹、斑丘疹和脓疱皮疹。
表 4 总结了 CodeBreaK 100 中观察到的选定实验室不良反应。
表 4. 在 CodeBreaK 100 中接受 LUMAKRAS 治疗的 KRAS G12C 突变 NSCLC 患者中较基线恶化的部分实验室异常 (≥ 20%)
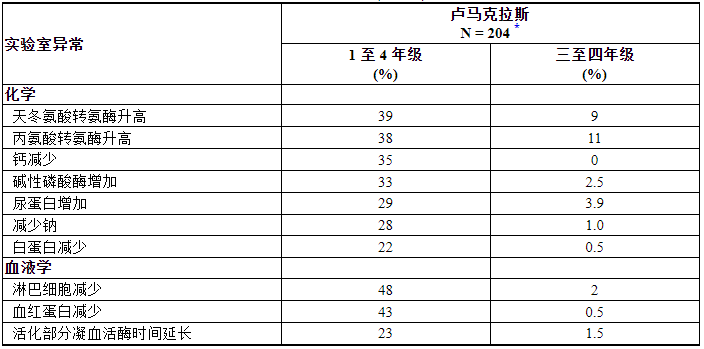
*N = 对感兴趣的参数至少进行过一次研究评估的患者数量。
7 药物相互作用
7.1 其他药物对 LUMAKRAS 的影响
酸还原剂
sotorasib的溶解度取决于 pH 值。LUMAKRAS与胃酸减少剂共同给药降低sotorasib浓度[见临床药理学(12.3) ] ,这可能降低sotorasib的功效。避免 LUMAKRAS 与质子泵抑制剂 (PPI)、H 2受体拮抗剂和局部作用抗酸剂共同给药。如果无法避免与酸还原剂共同给药,在局部作用抗酸剂给药前4小时或给药后10小时给予LUMAKRAS [见剂量和给药方法(2.4) ]。
强效 CYP3A4 诱导剂
Sotorasib是 CYP3A4 底物。LUMAKRAS与强CYP3A4诱导剂的共同给药降低sotorasib浓度[见临床药理学(12.3) ] ,这可能降低sotorasib的功效。避免 LUMAKRAS 与强 CYP3A4 诱导剂共同给药。
7.2 LUMAKRAS 对其他药物的影响
CYP3A4 底物
Sotorasib是一种 CYP3A4 诱导剂。LUMAKRAS与CYP3A4底物的共同给药降低其血浆浓度[见临床药理学(12.3) ],这可能降低底物的功效。避免 LUMAKRAS 与 CYP3A4 敏感底物共同给药,因为最小浓度变化可能导致底物治疗失败。如果无法避免共同给药,请根据其处方信息增加敏感 CYP3A4 底物剂量。
P-糖蛋白 (P-gp) 底物
Sotorasib是一种 P-gp 抑制剂。LUMAKRAS与P-gp底物共同给药增加其血浆浓度[见临床药理学(12.3) ],这可能增加底物的不良反应。避免 LUMAKRAS 与 P-gp 底物共同给药,因为最小的浓度变化可能会导致严重的毒性。如果无法避免联合给药,请根据其处方信息减少 P-gp 底物剂量。
乳腺癌抗性蛋白 (BCRP) 底物
Sotorasib是一种 BCRP 抑制剂。LUMAKRAS与BCRP底物共同给药增加其血浆浓度[见临床药理学(12.3) ],这可能增加底物不良反应的风险。当与 LUMAKRAS 共同给药时,监测 BCRP 底物的不良反应并根据其处方信息减少 BCRP 底物剂量。
8 在特定人群中的使用
8.1 怀孕
风险总结
没有关于孕妇使用 LUMAKRAS 的可用数据。在大鼠和兔胚胎-胎儿发育研究中,口服sotorasib在暴露量高达 960 mg 临床剂量人体暴露量的 4.6 倍时不会引起不良发育影响或胚胎致死性(参见数据)。
在美国普通人群中,临床认可的妊娠中重大出生缺陷和流产的背景风险估计分别为 2% 至 4% 和 15% 至 20%。
数据
动物数据
在一项大鼠胚胎-胎儿发育研究中,在器官形成期间对怀孕大鼠每日一次口服 sotorasib 导致母体毒性,剂量水平为 540 mg/kg(根据曲线下面积(AUC,约为人体暴露量的 4.6倍) ) 临床剂量为 960 mg)。Sotorasib在剂量高达 540 mg/kg 时不会引起不良发育影响,也不影响胚胎-胎儿存活。
在一项兔胚胎-胎儿发育研究中,在器官形成期每天口服一次 sotorasib,在 100 mg/kg 剂量水平(约 2.6 倍)可降低胎儿体重,并减少胎儿骨化掌骨数量。人体暴露量(基于临床剂量 960 mg 的 AUC),与母体毒性相关,包括给药阶段体重增加和食物消耗减少。Sotorasib在剂量高达 100 mg/kg 时不会引起不良发育影响,也不影响胚胎-胎儿存活。
8.2 哺乳期
风险总结
没有关于母乳中是否存在sotorasib或其代谢物、对母乳喂养儿童或产奶量的影响的数据。由于母乳喂养的儿童可能会出现严重不良反应,建议女性在 LUMAKRAS 治疗期间以及最终剂量后 1 周内不要母乳喂养。
8.4 儿童使用
LUMAKRAS 在儿童患者中的安全性和有效性尚未确定。
8.5 老年人使用
在 CodeBreaK 100 中,每天一次口服 LUMAKRAS 960 mg 的 357 名任何肿瘤类型患者中,46% 为 65 岁及以上,10% 为 75 岁及以上。老年患者和年轻患者之间没有观察到安全性或有效性的总体差异。
8.6 肝损伤
对于轻度至中度肝功能不全(Child Pugh A 或 B)的患者,不建议调整剂量。
严重肝损伤(Child-Pugh C)对 LUMAKRAS 安全性的影响尚不清楚。更频繁地监测有肝受损患者的sotorasib不良反应,因为这些患者可能出现不良反应包括肝毒性的风险增加[见临床药理学(12.3) ]。
11 描述
Sotorasib是 RAS GTPase 家族的抑制剂。分子式为C 30 H 30 F 2 N 6 O 3,分子量为560.6g/mol。sotorasib的化学名称是6-氟-7-(2-氟-6-羟基苯基)-(1M)-1-[4-甲基-2-(丙-2-基)吡啶-3-基]-4 -[(2S)-2-甲基-4-(丙-2-烯酰基)哌嗪-1-基]吡啶并[2,3-d]嘧啶-2(1H)-酮。
sotorasib的化学结构如下所示:
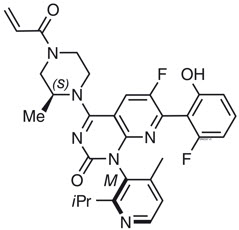
Sotorasib的 pKa 值为 8.06 和 4.56。sotorasib在水介质中的溶解度在 pH 1.2 至 6.8 范围内从 1.3 mg/mL 降低至 0.03 mg/mL。
LUMAKRAS 以口服薄膜衣片形式提供,含有 320 mg 或 120 mg sotorasib。片芯中的非活性成分是微晶纤维素、一水乳糖、交联羧甲基纤维素钠和硬脂酸镁。薄膜包衣材料由聚乙烯醇、二氧化钛、聚乙二醇、滑石粉、氧化铁黄和氧化铁红组成(仅限320毫克片剂)。
12 临床药理学
12.1 作用机制
Sotorasib是 KRAS G12C的抑制剂,KRAS G12C 是 RAS GTPase KRAS 的肿瘤限制性突变致癌形式。Sotorasib与 KRAS G12C的独特半胱氨酸形成不可逆的共价键,将蛋白质锁定在非活性状态,从而阻止下游信号传导而不影响野生型 KRAS。Sotorasib仅在KRAS G12C肿瘤细胞系中阻断 KRAS 信号传导、抑制细胞生长并促进细胞凋亡。Sotorasib在体外和体内抑制 KRAS G12C,且可检测到的脱靶活性极低。在小鼠肿瘤异种移植模型中,sotorasib-治疗导致肿瘤消退并延长生存期,并且与KRAS G12C模型中的抗肿瘤免疫相关。
12.2 药效学
Sotorasib暴露-反应关系和药效学反应的时间进程尚不清楚。
心脏电生理学
在批准的推荐剂量下,LUMAKRAS 不会导致 QTc 间期平均大幅增加(> 20 毫秒)。
12.3 药代动力学
sotorasib的药代动力学已在健康受试者和KRAS G12C突变实体瘤(包括 NSCLC)患者中进行了表征。Sotorasib在每天一次 180 mg 至 960 mg(批准推荐剂量的 0.19 至 1 倍)的剂量范围内表现出非线性、时间依赖性药代动力学,各剂量之间具有相似的全身暴露量(即 AUC 0-24h和C max)处于稳定状态。薄膜包衣片剂和预分散于水中的薄膜包衣片剂在空腹条件下给药时Sotorasib的全身暴露量相当。索托拉西卜血浆浓度在22天内达到稳定状态。重复给予 LUMAKRAS 剂量后未观察到蓄积,平均蓄积率为 0.56(变异系数 (CV):59%)。
吸收
sotorasib达到血浆峰浓度的中位时间为 1 小时。
食物的影响
当患者给予960 mg LUMAKRAS与高脂肪、高热量膳食(含有约800至1000卡路里,其中150、250和500至600卡路里分别来自蛋白质、碳水化合物和脂肪)时,sotorasib AUC 0-24h与空腹条件下给药相比增加了 25%。
分配
sotorasib稳态时的平均分布容积 (V d )为 211 L (CV: 135%)。在体外,sotorasib血浆蛋白结合率为 89%。
消除
sotorasib平均终末消除半衰期为 5 小时(标准差 (SD):2)。每天一次 960 mg LUMAKRAS 时,sotorasib稳态表观清除率为 26.2 L/hr (CV: 76%)。
代谢
sotorasib的主要代谢途径是非酶结合和与CYP3As的氧化代谢。
排泄
单剂量放射性标记的sotorasib后,74% 的剂量在粪便中回收(53% 不变),6%(1% 不变)在尿液中回收。
特定人群
根据年龄(28至86岁)、性别、种族(白人、黑人和亚洲人)、体重(36.8至157.9 kg)、治疗线、ECOG PS(0, 1)、轻度和中度肾功能损害(eGFR:≥30 mL/min/1.73 m 2),或轻度肝功能损害(AST或ALT < 2.5 × ULN或总胆红素< 1.5 × ULN)。尚未研究严重肾功能损害对sotorasib药代动力学的影响。
肝损伤
与肝功能正常受试者相比,单剂量索托拉西布的平均 AUC在中度肝受损受试者 (Child-Pugh B) 中降低 25%,在重度肝受损受试者 (Child-Pugh C) 中增加 4% 960 毫克 LUMAKRAS。
药物相互作用研究
临床研究
酸还原剂:重复剂量奥美拉唑(PPI)与单剂量LUMAKRAS共同给药在进食条件下降低sotorasib C max 65%和AUC 57%,并且在进食条件下降低sotorasib C max 57%和AUC 42%禁食条件。在进食条件下单剂量LUMAKRAS之前10小时和之后2小时给予单剂量法莫替丁(H 2受体拮抗剂)共同给药降低sotorasib C max 35%和AUC 38% 。
强CYP3A4诱导剂:重复剂量利福平(一种强CYP3A4诱导剂)与单剂量LUMAKRAS共同给药使sotorasib C max降低35%和AUC降低51%。
其他药物:LUMAKRAS 与伊曲康唑(一种强 CYP3A4 和 P-gp 联合抑制剂)和单剂量利福平(一种 OATP1B1/1B3 抑制剂)或二甲双胍(一种 MATE1)共同给药后,未观察到对 sotorasib 暴露有临床意义的影响。/MATE2-K 基板)。
CYP3A4 底物:LUMAKRAS 与咪达唑仑(一种敏感的 CYP3A4 底物)共同给药使咪达唑仑 C max降低48%,AUC 降低 53%。
P-gp 底物:LUMAKRAS 与地高辛(一种 P-gp 底物)共同给药可使地高辛 C max增加91%,AUC 增加 21%。
MATE1/MATE2-K 底物:与 LUMAKRAS 共同给药后,未观察到对二甲双胍(MATE1/MATE2-K 底物)暴露没有临床意义的影响。
BCRP 底物: LUMAKRAS 与瑞舒伐他汀(BCRP 底物)共同给药使瑞舒伐他汀 C max增加70%,AUC 增加 34%。
体外研究
细胞色素 P450 (CYP)酶:Sotorasib可能诱导 CYP2C8、CYP2C9 和 CYP2B6。Sotorasib不抑制 CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19 或 CYP2D6。
13 非临床毒理学
13.1 致癌、突变、生育能力受损
尚未对sotorasib进行致癌性研究。
Sotorasib在体外细菌回复突变 (Ames) 试验中不具有致突变性,并且在体内大鼠微核和彗星试验中不具有遗传毒性。
未使用sotorasib进行生育力/早期胚胎发育研究。在狗和大鼠中进行的一般毒理学研究没有对雌性或雄性生殖器官产生不良影响。
13.2 动物毒理学和/或药理学
在大鼠中,肾毒性包括轻微至显着的组织学肾小管变性/坏死以及肾脏重量、尿素氮、肌酐和肾小管损伤的尿生物标志物的增加,其剂量导致暴露约≥0.5倍于人临床剂量下的AUC。 960 毫克。与人类相比,大鼠肾脏中半胱氨酸 S-缀合物 β-裂解酶途径代谢的增加可能使大鼠比人类更容易受到肾毒性的影响,因为局部形成了推定的含硫代谢物。
在狗身上进行的为期 3 个月的毒理学研究中,sotorasib引起肝脏(小叶中心肝细胞肥大)、垂体(嗜碱性粒细胞肥大)和甲状腺(显着的滤泡细胞萎缩、中度至显着的胶体耗竭和滤泡细胞肥大)的发现根据临床剂量 960 mg 的 AUC,暴露量约为人体暴露量的 0.4 倍。这些发现可能是由于对肝细胞酶诱导的适应性反应以及随后的甲状腺激素水平降低(即继发性甲状腺功能减退症)所致。虽然没有测量狗的甲状腺水平,但在体外狗肝细胞测定中证实了已知参与甲状腺激素代谢的尿苷二磷酸葡萄糖醛酸基转移酶的诱导。
14 临床研究
LUMAKRAS 的疗效在一项单臂、开放标签、多中心试验 (CodeBreaK 100 [NCT03600883]) 中入组的一部分患者中得到了证实。符合条件的患者必须患有局部晚期或转移性KRAS G12C突变 NSCLC,且在接受免疫检查点抑制剂和/或铂类化疗后疾病进展,东部肿瘤合作组表现状态 (ECOG PS) 为 0 或 1,并且至少一个实体瘤疗效评估标准 (RECIST v1.1) 定义的可测量病变。
所有患者都需要使用在中心实验室进行的QIAGEN therascreen ® KRAS RGQ PCR 试剂盒前瞻性地鉴定肿瘤组织样本中的KRAS G12C突变 NSCLC。在总共 126 名入组受试者中,有 2 名受试者 (2%) 由于基线时缺乏放射学可测量的病变而无法进行疗效分析评估。在肿瘤组织中确认有KRAS G12C突变的 124 名患者中,使用 Guardant360 ® CDx 对 112 名患者的血浆样本进行了回顾性检测。78/112 名患者 (70%)在血浆样本中发现KRAS G12C突变,31/112 名患者 (28%) 没有KRAS G12C由于 Guardant360 ® CDx 测试失败,血浆样本中发现的突变和 3/112 (2%) 无法评估。
共有 124 名患者在基线时至少有一个可测量的病变,并根据 RECIST v1.1 进行盲法独立中央审查 (BICR) 评估,并接受 LUMAKRAS 960 mg 每日一次治疗,直至疾病进展或出现不可接受的毒性。主要疗效结果指标是客观缓解率 (ORR) 和缓解持续时间 (DOR),由 BICR 根据 RECIST v1.1 进行评估。
研究人群的基线人口统计学和疾病特征为: 中位年龄 64 岁(范围:37 至 80 岁),其中 48% ≥ 65 岁,8% ≥ 75 岁;50% 女性;82% 白人、15% 亚洲人、2% 黑人;70% ECOG PS 1;96% 患有 IV 期疾病;99% 为非鳞状组织学;81% 曾经吸烟,12% 当前吸烟,5% 从不吸烟。所有患者均接受过至少 1 种既往针对转移性 NSCLC 的全身治疗;43% 仅接受过 1 种既往治疗,35% 接受过 2 种既往治疗,23% 接受过 3 种既往治疗;91% 的患者既往接受过抗 PD-1/PD-L1 免疫治疗,90% 的患者接受过铂类化疗,81% 的患者同时接受过铂类化疗和抗 PD-1/PD-L1。已知的胸外转移部位包括 48% 骨、21% 脑和 21% 肝。
功效结果总结于表5中。
表 5. 在 CodeBreaK 100 中接受 LUMAKRAS 治疗的 KRAS G12C 突变 NSCLC 患者的疗效结果
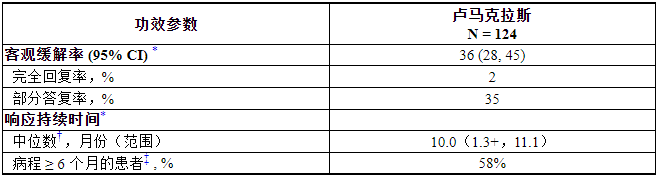
CI = 置信区间
*由盲法独立中央审查 (BICR) 评估
†使用 Kaplan-Meier 方法进行估计
‡观察到反应持续时间超过里程碑时间的患者比例
16 如何供应/储存和处理
如何供应
LUMAKRAS(sotorasib)320毫克片剂为米色,椭圆形,薄膜包衣,一侧凹陷有“AMG”,另一侧凹陷有“320”,供应如下:
- 纸盒装一瓶 90 片药片,带防儿童开启盖,NDC 55513-504-50
LUMAKRAS(sotorasib)120毫克片剂为黄色,长方形,薄膜包衣,一侧凹陷有“AMG”,另一侧凹陷有“120”,供应如下:
- 装有两瓶 120 片药片的纸箱,带防儿童开启盖,NDC 55513-488-02
- 纸盒装一瓶 240 片药片,带防儿童开启盖,NDC 55513-488-24
储存和处理
储存温度为 20°C 至 25°C(68°F 至 77°F)。允许温度范围为 15°C 至 30°C(59°F 至 86°F)[请参阅 USP 控制室温]。
17 患者咨询信息
建议患者阅读 FDA 批准的患者标签(患者信息)。
肝毒性
建议患者立即联系他们的医疗保健提供者了解肝功能障碍的体征和症状[见警告和注意事项(5.1) ]。
间质性肺疾病 (ILD)/肺炎
建议患者立即联系他们的医疗保健提供者报告新的或恶化的呼吸道症状[见警告和注意事项(5.2) ]。
哺乳期
建议妇女用LUMAKRAS治疗期间和最后剂量后1周不要母乳喂养[见特殊人群中使用(8.2) ]。
药物相互作用
建议患者告知其医疗保健提供者所有伴随药物,包括处方药、非处方药、维生素、饮食和草药产品。告知患者服用LUMAKRAS时避免质子泵抑制剂和H 2受体拮抗剂[见药物相互作用(7.1)和(7.2) ]。
如果无法避免与酸还原剂共同给药,告知患者在局部作用抗酸剂之前4小时或之后10小时服用LUMAKRAS [见剂量和给药方法(2.4) ]。
漏服剂量
如果LUMAKRAS错过剂量超过6小时,第二天按规定恢复治疗[见剂量和给药方法(2.2) ]。
制造商:
Amgen Inc.
One Amgen Center Drive
Hundred Oaks, CA 91320-1799 USA
专利:http://pat.amgen.com/lumakras/
© 2021、2022、2023 Amgen Inc. 保留所有权利。
1XXXXXX – V4

什么是 LUMAKRAS?
LUMAKRAS 是一种处方药,用于治疗成人非小细胞肺癌 (NSCLC):
- 已扩散到身体其他部位或无法通过手术切除,并且
- 其肿瘤具有异常的 KRAS G12C 基因,并且
- 之前至少接受过一种癌症治疗的人。
您的医疗保健提供者将进行测试,以确保 LUMAKRAS 适合您。
目前尚不清楚 LUMAKRAS 对儿童是否安全有效。
在服用 LUMAKRAS 之前我应该告诉我的医疗保健提供者什么?
在服用 LUMAKRAS 之前,请告诉您的医疗保健提供者您的所有医疗状况,包括您是否:
- 有肝脏问题。
- 有肺癌以外的肺部或呼吸问题。
- 已怀孕或计划怀孕。目前尚不清楚 LUMAKRAS 是否会伤害您未出生的婴儿。
- 正在母乳喂养或计划母乳喂养。目前尚不清楚 LUMAKRAS 是否会进入您的母乳。在 LUMAKRAS 治疗期间以及最后一次给药后 1 周内不要母乳喂养。
告诉您的医疗保健提供者您服用的所有药物,包括处方药和非处方药、维生素、膳食和草药补充剂。LUMAKRAS 可以影响某些其他药物的作用方式,而其他一些药物也可以影响 LUMAKRAS 的作用方式。如果您在 LUMAKRAS 治疗期间服用抗酸药物,包括质子泵抑制剂 (PPI) 药物或 H 2阻滞剂,请特别告知您的医疗保健提供者。如果您不确定,请询问您的医疗保健提供者。我应该如何服用 LUMAKRAS?
- 完全按照您的医疗保健提供者的指示服用 LUMAKRAS。除非您的医疗保健提供者告诉您,否则请勿改变您的剂量或停止服用 LUMAKRAS。
- 每天大约在同一时间服用处方剂量的 LUMAKRAS 1 次。
- 随餐或单独服用 LUMAKRAS 均可。
- 整个吞下 LUMAKRAS 药片。不要咀嚼、压碎或劈开药片。
- 如果您无法吞服整个 LUMAKRAS 药片:
- 将处方剂量的 LUMAKRAS 放入一杯 4 盎司(120 毫升)非碳酸室温水中,不要压碎药片。请勿使用任何其他液体。
- 搅拌或旋转杯子约 3 分钟,直至药片变成小块(药片不会完全溶解)。混合物的颜色可以是浅黄色至亮黄色。
- 立即或在准备后 2 小时内饮用 LUMAKRAS 和水的混合物。不要咀嚼药片碎片。
- 再用 4 盎司(120 毫升)水冲洗玻璃杯并饮用,以确保您服用了全剂量的 LUMAKRAS。
- 如果您不立即饮用混合物,请在饮用前再次搅拌或旋转混合物。
- 如果您服用抗酸药,请在服用抗酸药前 4 小时或服用后 10 小时服用 LUMAKRAS。
- 如果您错过了一剂 LUMAKRAS,请在想起来后立即补服。如果已经超过 6 小时,请勿服用该剂量。第二天在您定期安排的时间服用下一剂。不要同时服用 2 剂以弥补错过的剂量。
- 如果您在服用一剂 LUMAKRAS 后呕吐,请勿服用额外剂量。第二天在您定期安排的时间服用下一剂。
LUMAKRAS 可能有哪些副作用?
LUMAKRAS 可能会引起严重的副作用,包括:
- 肝脏问题。LUMAKRAS 可能会导致肝脏血液检查结果异常。您的医疗保健提供者应在开始使用 LUMAKRAS 之前和治疗期间进行血液检查,以检查您的肝功能。如果您出现任何肝脏问题的体征或症状,请立即告诉您的医疗保健提供者,包括:
- 您的皮肤或眼睛的白色部分变黄(黄疸)
- 深色或“茶色”尿液
- 浅色粪便(排便)
- 疲倦或虚弱
- 恶心或呕吐
- 出血或瘀伤
- 食欲不振
- 胃部右侧(腹部)疼痛、疼痛或压痛
- 肺部或呼吸问题。LUMAKRAS 可能会引起肺部炎症,从而导致死亡。如果您出现新的呼吸急促、咳嗽或发烧症状或症状加重,请立即告诉您的医疗保健提供者或寻求紧急医疗帮助。
如果您出现副作用,您的医疗保健提供者可能会改变您的剂量、暂时停止或永久停止 LUMAKRAS 治疗。
LUMAKRAS 最常见的副作用包括:
- 腹泻
- 肌肉或骨骼疼痛
- 恶心
- 疲倦
- 肝脏问题
- 咳嗽
- 肝功能检查的变化
- 某些其他血液检查的变化
这些并不是 LUMAKRAS 可能产生的所有副作用。
打电话给您的医生,征求有关副作用的医疗建议。您可以拨打 1-800-FDA-1088 向 FDA 报告副作用。
您还可以拨打 1-800-772-6436 (1-800-77-AMGEN) 向安进报告副作用。
我应该如何储存 LUMAKRAS?
- 将 LUMAKRAS 储存在 68°F 至 77°F(20°C 至 25°C)的室温下。
- 瓶子有一个防儿童开启的盖子。
将 LUMAKRAS 和所有药物放在儿童接触不到的地方。
有关安全有效使用 LUMAKRAS 的一般信息。
有时开出药物的目的并非患者信息传单中列出的目的。请勿将 LUMAKRAS 用于未处方的病症。不要将 LUMAKRAS 给予其他人,即使他们有与您相同的症状。这可能会伤害他们。您可以向您的医疗保健提供者或药剂师询问为医疗保健专业人员编写的有关 LUMAKRAS 的信息。
LUMAKRAS 的成分是什么?
- 活性成分: sotorasib
- 非活性成分:微晶纤维素、一水乳糖、交联羧甲基纤维素钠和硬脂酸镁。片剂薄膜包衣材料含有聚乙烯醇、二氧化钛、聚乙二醇、滑石粉、氧化铁黄和氧化铁红(仅限 320 毫克片剂)。
制造商:Amgen Inc., One Amgen Center Drive, Hundred Oaks, CA 91320-1799 USA
© 2021 Amgen Inc. 保留所有权利。
如需了解更多信息,请访问 www.LUMAKRAS.com 或致电 1-800-772-6436 (1-800-77-AMGEN)。
本患者信息已获得美国食品和药物管理局的批准。
修订日期:01/2023
[零件编号] v2
主要显示面板 - 120 毫克片剂瓶纸箱标签
NDC 55513-488-24
LUMAKRAS™
(sotorasib)片剂
120毫克
每片含有 120 毫克sotorasib。
储存温度为 20°C 至 25°C(68°F 至 77°F)。
允许温度范围为 15°C 至 30°C
(59°F 至 86°F)。
推荐剂量:参见处方
信息。
安进®
主要显示面板 - 320 毫克片剂瓶纸箱标签
NDC 55513-504-50
LUMAKRAS® (
sotorasib )片剂
320 毫克 | 新力量
每片含有 320 毫克sotorasib。
储存温度为 20°C 至 25°C(68°F 至 77°F)。
允许温度范围为 15°C 至 30°C
(59°F 至 86°F)。
推荐剂量:参见处方
信息。
安进®

【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
本说明书来源于:美国FDA
https://nctr-crs.fda.gov/fdalabel/services/spl/set-ids/c80a362c-7ac3-4894-a076-0691e68ef8c1/spl-doc?hl=Sotorasib
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书





【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
-

-

-

-

通用名: 索托雷塞片
商品名: LUCISOT
规格: 120mg×56片
产地: 卢修斯医药(老挝)有限公司(LUCIUS PHARMACEUTICALS(LAO) CO.,LTD)
国际参考零售价:¥**/盒
-