商品详情
返回产品目录



商品包装及说明书因厂家更换频繁,如有不符以实物为主
注射用戈沙妥珠单抗
国际零售参考价:¥**/瓶
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。
警告:中性粒细胞减少症和腹泻
- 可能发生严重的中性粒细胞减少。扣压TRODELVY为低于1500个/ mm绝对中性粒细胞计数3 或中性粒细胞减少发热。在治疗期间定期监测血细胞计数。考虑使用G-CSF进行二级预防。立即对发热性中性粒细胞减少症患者进行抗感染治疗 [见警告和注意事项(5.1)]。
- 可能会出现严重腹泻。监测腹泻患者,并根据需要给予液体和电解质。如果没有禁忌,请服用阿托品以缓解任何严重程度的早期腹泻。在晚期腹泻发作时,应评估感染原因,如果为阴性,应立即启动洛哌丁胺 [见警告和注意事项(5.2)]。 如果发生严重腹泻,请暂不使用TRODELVY直至降至 < 1级并减少随后的剂量 [参见剂量和用法(2.3)]。
1适应症和用途
TRODELVY用于治疗转移性三阴性乳腺癌(mTNBC)的成年患者,这些患者已接受至少两种先前的转移性疾病疗法。
根据肿瘤反应率和反应持续时间,该适应症得到加速批准[见临床研究(14)]。对于该适应症的持续批准可能取决于验证试验中对临床益处的验证和描述。
2剂量和给药
2.1重要使用信息
请勿用TRODELVY替代或与含有伊立替康或其活性代谢产物SN-38的其他药物一起使用。
2.2推荐剂量和时间表
TRODELVY推荐剂量为10 mg / kg,在21天治疗周期的第1天和第8天每周一次静脉输注。继续治疗直至疾病进展或出现不可接受的毒性。请勿以大于10 mg / kg的剂量服用TRODELVY。
辖TRODELVY仅作为静脉内输注。请勿静脉推注或推注。
第一次输注:3个小时内进行输注。在输注过程中以及初始剂量后至少30分钟观察患者,以了解输注相关反应的体征或症状[请参阅警告和注意事项(5.3)]。
后续输液:如果可以耐受以前的输液,则应在1到2个小时内进行输液。在输液期间以及输液后至少30分钟观察患者。
服药前
在每次服用TRODELVY之前,建议进行预防输注反应和预防化疗引起的恶心和呕吐(CINV)的处方药。
- 输注前应先使用退热药,H1和H2阻滞剂进行预用药,皮质类固醇可用于先前有输注反应的患者。
- 具有两种或三种药物联合治疗方案的前药(例如,具有5-HT3受体拮抗剂或NK 1受体拮抗剂的地塞米松,以及所示的其他药物)。
2.3不良反应的剂量修改
输液相关反应
如果患者出现与输注相关的反应,请减慢或中断TRODELVY的输注速度。永久终止TRODELVY以进行危及生命的输液相关反应[请参阅警告和注意事项(5.3)]
不良反应的剂量调整
如表1 所示,停用或终止TRODELVY来管理不良反应。减少不良反应的剂量后,请勿重新增加TRODELVY的剂量。
表1:不良反应的剂量修改

2.4管理准备
重组
- TRODELVY是一种细胞毒性药物。
- 遵循适用的特殊处理和处置程序1。
- 在每个治疗周期开始时根据患者体重计算TRODELVY所需剂量(mg)(如果自上次给药后患者体重变化超过10%,则更频繁)[请参阅剂量和给药方法(2.1) ]。
- 让所需数量的小瓶加热到室温。
- 使用无菌注射器将20 mL的0.9%氯化钠注射液USP缓慢注入每个180 mg TRODELVY小瓶中。所得浓度将为10 mg / mL。
- 轻轻旋转小瓶,最多溶解15分钟。不要摇晃。只要溶液和容器允许,在给药前应目视检查肠胃外药品中是否有颗粒物和变色。溶液应不含可见的颗粒,透明和黄色。如果溶液变混浊或变色,请勿使用。
- 立即使用以准备稀释的TRODELVY输液。
稀释
- 根据患者的体重计算获得适当剂量所需的TROTROVY溶液的体积。使用注射器从小瓶中取出此量。丢弃小瓶中剩余的所有未使用部分。
- 将所需体积的TRODELVY溶液慢慢注入聚丙烯(PP)输液袋中,以最大程度地减少泡沫。请勿摇动内容。
- 根据需要,使用0.9%氯化钠注射液(USP)调整输液袋中的体积,以获得1.1 mg / mL至3.4 mg / mL的浓度(总体积不应超过500 mL)。对于体重超过170千克的患者,将TRODELVY的总剂量平均分配在两个500 mL输液袋之间,并通过缓慢输注顺序输注。
- 只能使用0.9%的氯化钠注射液(USP),因为尚未用其他基于输液的溶液确定重构产品的稳定性。立即在输液袋中使用稀释的溶液。如果不立即使用,装有TRODELVY溶液的输液袋可在2°C至8°C(36°F至46°F)的冷藏条件下最多保存4小时。冷藏后,在4小时内(包括输注时间)施用稀释液。
请勿冻结或摇动。避光。
行政
- 将TRODELVY静脉内输注。保护输液袋避光。
- 可以使用输液泵。
- 请勿将TRODELVY或与其他药品一起输注使用。
- 输液完成后,用20 mL 0.9%氯化钠注射液USP冲洗静脉管线。
3剂型和强度
注射剂:单剂量小瓶中180 mg灰白色至淡黄色冻干粉末。
4禁忌症
TRODELVY是对TRODELVY 发生严重超敏反应的患者的禁忌症[请参阅警告和注意事项(5.3)]。
5警告和注意事项
5.1中性粒细胞减少
TRODELVY可引起严重或危及生命的中性粒细胞减少症。扣压TRODELVY为低于1500个/ mm绝对中性粒细胞计数3上低于1000个/ mm任何周期或中性粒细胞计数的第1天3上的任何周期的第8天。扣压TRODELVY为中性粒细胞减少发热。由于中性粒细胞减少症,可能需要调整剂量[见剂量和用法(2.3)]。
发生在6%中性粒细胞减少性发热(408分之24)患者治疗TRODELVY,包括8%(108分之9)患者mTNBC至少两个先前治疗之后。不到1%(1/408)的患者出现发热性中性粒细胞减少症,导致永久性停药。
mTNBC患者中1-4级中性粒细胞减少的发生率为64%(n = 108)。在所有接受TRODELVY治疗的患者中(n = 408),1-4级中性粒细胞减少的发生率为54%。4级中性粒细胞减少症发生率为13%。少于1%(2/408)的患者因中性粒细胞减少症而永久终止治疗。
5.2腹泻
TRODELVY可引起严重的腹泻。扣压TRODELVY在预定的治疗管理和恢复的时间3-4级腹泻时解析为≤1级[ 见剂量和给药方法(2.3) ]。
腹泻开始时,应评估感染原因,如果为阴性,应立即开始洛哌丁胺治疗,最初为4毫克,随后每次腹泻为2毫克,每天最多腹泻16毫克。腹泻缓解后12小时停止使用洛哌丁胺。如临床指示,还可以采用其他支持措施(例如,补充液体和电解质)。
对TRODELVY治疗表现出过多胆碱能反应的患者(例如腹部绞痛,腹泻,流涎等),可以接受适当的前药(例如阿托品)进行后续治疗。
接受TRODELVY治疗的mTNBC患者中有63%(68/108)发生腹泻,所有患者中有62%(254/408)发生腹泻。在每个人口中,3-4级的事件发生在mTNBC患者的9%(108分之10)中,并用所有患者的9%(408分之36)TRODELVY。408名患者中有4名(<1%)因腹泻而中止治疗。在mTNBC队列中,有2%(2/108)的患者中有中性粒细胞减少性结肠炎,在接受TRODELVY治疗的所有患者中有1%。
5.3过敏
TRODELVY可能导致严重的威胁生命的超敏反应。TRODELVY在临床试验中已观察到过敏反应。
用TRODELVY治疗的患者中有37%(151/408)在给药后24小时内出现超敏反应。3-4级过敏发生的与治疗的患者的1%(408分之6)TRODELVY。导致TRODELVY永久终止的超敏反应发生率为1%(3/408)。
建议接受TRODELVY治疗的患者进行输液前药物治疗。在每次TRODELVY输注期间以及完成每次输注后至少30分钟内,密切观察患者的输注相关反应[参见剂量和用法(2.3)]。治疗此类反应的药物以及应急设备应可立即使用。
5.4恶心和呕吐
TRODELVY具有催吐作用。恶心的发生率为69%(108分之74)与mTNBC与治疗所有患者的69%(408分之281)TRODELVY。在这些人群中,分别有6%(7/108)和5%(22/408)发生了3级恶心。
呕吐的患者发生49%(53/108)与mTNBC并用所有患者的45%(408分之183)TRODELVY。这些患者中分别有6%(7/108)和4%(16/408)发生3级呕吐。
预先用两种或三种药物联合用药(例如,带有5-HT3受体拮抗剂或NK-1受体拮抗剂的地塞米松以及所示的其他药物)预防化疗引起的恶心和呕吐(CINV)。
在计划的治疗给药时,对于3级恶心或3-4级呕吐应停用TRODELVY剂量,当分辨率降至≤1级时,应继续采取其他支持措施[见剂量和给药方法(2.3)]。
临床上也可采用其他止吐药和其他支持措施。应给所有患者带回家的药物,并有明确的预防和治疗恶心和呕吐说明。
5.5用于UGT1A1活性降低的患者
尿苷二磷酸-葡萄糖醛酸转移酶1A1(UGT1A1)* 28等位基因纯合的个体患中性粒细胞减少症的风险增加,并且在开始TRODELVY治疗后可能面临其他不良反应的风险增加。
在84%(343/408)的患者中接受TRODELVY(在21天周期的第1天和第8 天时达到10 mg / kg)并具有回顾性UGT1A1基因型结果可用,4级中性粒细胞减少的发生率为26%( UGT1A1 * 28等位基因纯合的患者为10/39),UGT1A1 * 28等位基因杂合的患者为13%(20/155),野生型等位基因纯合的患者为11%(16/149) [请参见临床药理学(12.5)]。
密切监测UGT1A1活性降低的患者的严重中性粒细胞减少症。对于UGT1A1 * 28纯合子患者,适当剂量尚不明确,应根据患者对治疗的耐受性来考虑[见剂量和用法(2.3)]。
5.6胚胎-胎儿毒性
根据其作用机理,TRODELVY对孕妇给药可引起致畸性和/或胚胎-胎儿致死性。TRODELVY包含遗传毒性成分SN-38,并靶向快速分裂的细胞[请参见临床药理学(12.1)和非临床毒理学(13.1)]。建议孕妇和具有生殖潜力的女性对胎儿的潜在危险。劝告有生殖潜力的女性在TRODELVY治疗期间以及最后一次服药后的6个月内使用有效的避孕方法。建议具有生殖潜力的女性伴侣的男性患者在TRODELVY治疗期间使用有效的避孕方法和最后一次给药后3个月[参见特殊人群中使用(8.1,8.3)]。
6不良反应
标签的其他部分详细讨论了以下不良反应:
- 中性粒细胞减少症[请参阅警告和注意事项(5.1)]
- 腹泻[参见警告和注意事项(5.2)]
- 过敏[请参阅警告和注意事项(5.3)]
- 恶心和呕吐[请参阅警告和注意事项(5.4)]
6.1临床试验经验
由于临床试验是在广泛不同的条件下进行的,因此无法将在一种药物的临床试验中观察到的不良反应率直接与另一种药物在临床试验中观察到的不良反应率进行比较,并且可能无法反映在临床实践中观察到的不良反应率。
“警告和注意事项”部分中描述的数据反映了单臂开放标签研究(IMMU-132-01)中单药暴露于TRODELVY的情况,该研究针对408例mTNBC和其他恶性肿瘤患者接受了先前的全身性治疗方案晚期疾病。 在21天治疗周期的第1天和第8天,每周一次以TRODELVY静脉输注的方式给药,剂量最高为10 mg / kg,直到疾病进展或出现不可接受的毒性。
在数据表2反映暴露于TRODELVY中的108例mTNBC谁曾在研究接收的至少两个在先治疗转移性疾病(IMMU-132-01)的子集。患者在21天治疗周期的第1天和第8天通过静脉输注接受TRODELVY 10 mg / kg,直到疾病进展或出现不可接受的毒性。这108名患者的中位治疗持续时间为5.1个月(范围:0-51个月)。
据报道31%的患者出现严重的不良反应。接受TRODELVY治疗的患者中最常见的严重不良反应(报道> 1%)是发热性中性粒细胞减少(6%)呕吐(5%),恶心(3%),呼吸困难(3%),腹泻(4%),贫血(2%),胸腔积液,中性粒细胞减少,肺炎,脱水(各2%)。
TRODELVY因2%的患者不良反应而永久停药。导致停药的不良反应为过敏反应,厌食/疲劳,头痛(每例<1%,每例1名患者)。百分之四十五(45%)的患者出现不良反应,导致治疗中断。导致治疗中断的最常见不良反应是中性粒细胞减少(33%)。TRODELVY治疗的患者中有33%发生了导致剂量降低的不良反应,其中24%降低了一种剂量,9%降低了两种剂量。导致剂量减少的最常见不良反应是中性粒细胞减少/发热性中性粒细胞减少。
表2总结了IMMU-132-01研究中≥10%的mTNBC患者发生的不良反应。
表2:IMMU-132-01中≥10%的mTNBC患者的不良反应


表3:在接受TRODELVY治疗时,> 10%的患者观察到实验室异常

6.2免疫原性
与所有治疗性蛋白质一样,具有免疫原性的潜力。抗体形成的检测高度依赖于测定的灵敏度和特异性。另外,在测定中观察到的抗体(包括中和抗体)阳性的发生率可能受到多种因素的影响,包括测定方法,样品处理,样品收集的时间,伴随用药和基础疾病。由于这些原因,将以下所述研究中的抗体发生率与其他研究中或其他sacituzumab govitecan产品的抗体发生率进行比较可能会产生误导。
使用基于化学发光(ECL)的免疫测定法评估106例mTNBC患者血清中TRODELVY的免疫原性分析,以测试抗sacituzumab govitecan-hziy抗体。抗sacituzumab govitecan-hziy抗体的检测使用3层方法进行:筛选,确认和滴定度。在2%(2/106)的患者中出现了持久的抗sacituzumab govitecan-hziy抗体。
7药物相互作用
7.1其他药物对TRODELVY的影响
UGT1A1抑制剂
同时给予TRODELVY与UGT1A1的抑制剂可能增加不良反应的发生率是由于全身性暴露于SN-38的潜在增加[参见警告和注意事项(5.5)和临床药理学(12.3,12.5)]。避免与TRODELVY一起使用UGT1A1抑制剂。
UGT1A1诱导剂
暴露于SN-38可以在患者中显着减少伴随接收UGT1A1酶诱导剂[见警告和注意事项(5.5)和临床药理学(12.3,12.5)]。避免与TRODELVY一起使用UGT1A1诱导剂。
8在特定人群中的使用
8.1怀孕
风险摘要
根据其作用机理,TRODELVY对孕妇给药可引起致畸性和/或胚胎-胎儿致死性。孕妇没有可用的数据来告知与药物相关的风险。TRODELVY含有遗传毒性成分SN-38,对快速分裂的细胞有毒[参见临床药理学(12.1)和非临床毒理学(13.1)]。建议孕妇和具有生殖潜力的女性对胎儿的潜在危险。
对于指定人群,主要出生缺陷和流产的估计背景风险尚不清楚。在美国普通人群中,临床公认的怀孕中主要先天缺陷和流产的估计背景风险分别为2%至4%和15%至20%。
数据
动物资料
没有使用sacituzumab govitecan-hziy进行的生殖和发育毒理学研究。
8.2哺乳
风险摘要
没有关于人乳中存在sacituzumab govitecan-hziy或SN-38,对母乳喂养的孩子的影响或对牛奶生产的影响的信息。由于母乳喂养的孩子可能会出现严重的不良反应,因此建议妇女在治疗期间和最后一次服用TRODELVY后1个月内不要母乳喂养。
8.3生殖潜力的雌雄
验孕
在开始TRODELVY之前验证具有生殖潜能的女性的妊娠状况。
避孕
女性
向孕妇服用TRODELVY可能会造成胎儿伤害[见在特定人群中使用(8.1)]。劝告有生殖潜力的女性在TRODELVY治疗期间以及最后一次服药后的6个月内使用有效的避孕方法。
雄性
由于有潜在的遗传毒性,建议具有生殖潜力的女性伴侣的男性患者在TRODELVY治疗期间以及最后一次给药后的3个月内使用有效的避孕方法。
不孕症
女性
根据动物研究结果,TRODELVY可能会损害具有生殖潜能的雌性的生育力[见非临床毒理学(13.1)]。
8.4小儿使用
TRODELVY的安全性和有效性尚未在儿科患者中确立。
8.5老年用途
在接受TRODELVY治疗的患者中,有19/108名(18%)mTNBC患者和144/408名(35%)患者≥65岁。这些患者和年轻患者之间在安全性和有效性方面未观察到总体差异。
8.6肝功能不全
对轻度肝功能不全(胆红素小于或等于1.5 ULN且AST / ALT <3 ULN)的患者服用TRODELVY时,无需调整起始剂量。
轻度肝功能不全(胆红素小于或等于ULN且AST大于ULN,或胆红素大于1.0至1.5的ULN和AST的任何水平; n = 12)患者中TRODELVY的暴露与肝功能正常的患者相似(胆红素或AST小于ULN; n = 45)。
TRODELVY在中度或重度肝功能不全患者中的安全性尚未确立。TRODELVY尚未在血清胆红素> 1.5 ULN或AST和ALT> 3 ULN或AST和ALT> 5 ULN并伴有肝转移的患者中进行测试。
没有专门的试验来研究TRODELVY在中度或重度肝功能不全患者中的耐受性。对于这些患者的起始剂量没有建议。
10过量
在一项临床试验中,TRODELVY的计划剂量最高为18 mg / kg(约为最大推荐剂量10 mg / kg的1.8倍)。在这些患者中,观察到严重中性粒细胞减少症的发生率更高。
11说明
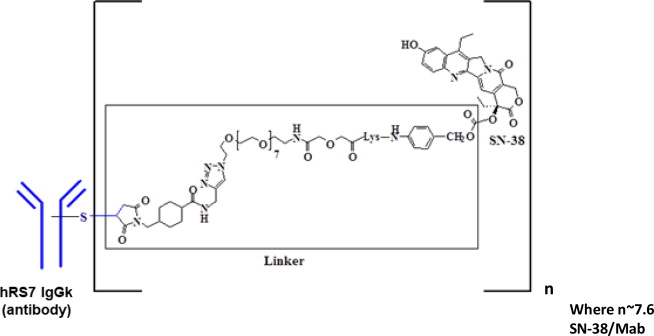
Sacituzumab govitecan-hziy是Trop-2定向抗体和拓扑异构酶抑制剂的结合物,由以下三个成分组成:
- 人源化单克隆抗体hRS7 IgG1ϰ(也称为sacituzumab),与Trop-2(滋养层细胞表面抗原2)结合。
- 药物SN-38,一种拓扑异构酶抑制剂;
- 一个可水解的接头(称为CL2A),可将人源化单克隆抗体与SN-38相连。
重组单克隆抗体是由哺乳动物(鼠骨髓瘤)细胞产生的,而小分子成分SN-38和CL2A是通过化学合成产生的。每个抗体分子中Sagovuzumab govitecan-hziy平均含有7至8个SN-38分子。曲妥珠单抗govitecan-hziy的分子量约为160千道尔顿。戈维替卡沙星单抗具有以下化学结构。

TRODELVY(sacituzumab govitecan-hziy)注射剂是无菌,无防腐剂,灰白色至淡黄色的冻干粉末,可在50 mL透明玻璃单剂量小瓶中静脉注射,并带有橡胶塞和铝盖扣封口脱帽。
每个TRODELVY单剂量小瓶可递送180毫克sacituzumab govitecan-hziy,77.3毫克2-(N-吗啉代)乙磺酸(MES),1.8毫克聚山梨酸酯80和154毫克海藻糖二水合物。用20 mL的0.9%氯化钠注射液(USP)复溶,结果浓度为10 mg / mL,pH值为6.5。
12临床药理学
12.1行动机制
Sacituzumab govitecan-hziy是Trop-2导向的抗体-药物偶联物。Sacituzumab是可识别Trop-2的人源化抗体。小分子SN-38是拓扑异构酶I抑制剂,它通过接头共价附于抗体。药理学数据表明,sacituzumab govitecan-hziy与表达Trop-2的癌细胞结合,并在随后通过接头水解释放SN-38时被内在化。SN-38与拓扑异构酶I相互作用并阻止拓扑异构酶I诱导的单链断裂的重新连接。产生的DNA损伤导致凋亡和细胞死亡。在三阴性乳腺癌小鼠异种移植模型中,Sacituzumab govitecan-hziy降低了肿瘤的生长。
12.2药效学
沙曲单抗govitecan-hziy的暴露-反应关系和药效学反应的时间过程未知。
12.3药代动力学
在一项以10 mg / kg剂量接受sacituzumab govitecan-hziy作为单药的mTNBC患者人群中的一项研究中,评估了sacituzumab govitecan-hziy和SN-38的血清药代动力学。表4列出了sacituzumab govitecan-hziy和游离SN-38的药代动力学参数。
表4:Sacituzumab Govitecan-hziy和游离SN-38的平均PK参数(±标准偏差)汇总

分配
sacituzumab govitecan-hziy的平均分配量为0.045 L / kg。
消除
sacituzumab govitecan-hziy和游离SN-38的平均半衰期分别为16小时和18小时。sacituzumab govitecan-hziy的清除率为0.002 L / h / kg。
代谢
尚无使用sacituzumab govitecan-hziy进行代谢研究。SN-38(sagovuzumab govitecan-hziy的小分子部分)通过UGT1A1代谢。在患者血清中可检测到SN-38的葡萄糖醛酸代谢产物(SN-38G)。
特定人群
在数量有限的mTNBC患者中进行药代动力学分析(n = 57),未发现年龄或种族对地瓜舒缓单抗药代动力学的影响。已知肾脏消除对SN-38的排泄的贡献最小,SN-38是sacituzumab govitecan-hziy的小分子部分。尚无肾功能不全或终末期肾脏疾病(CLcr≤30 mL / min)患者体内sacituzumab govitecan-hziy的药代动力学数据。
在轻度肝功能不全(胆红素小于或等于ULN且AST大于ULN,或胆红素大于1.0至小于1.5的ULN和AST的任何水平; n = 12)的患者中,sacituzumab govitecan-hziy的暴露相似肝功能正常的患者(胆红素或AST低于ULN; n = 45)。
中度或重度肝功能不全患者未知Sacituzumab govitecan-hziy暴露。由于肝UGT1A1活性降低,此类患者的SN-38暴露水平可能升高。
药物相互作用研究
尚无使用sacituzumab govitecan-hziy或其成分进行药物相互作用的研究,预计UGT1A1的抑制剂或诱导剂分别增加或减少SN-38的暴露[见药物相互作用(7)]。
12.5药物基因组学
SN-38通过UGT1A1代谢[ 参见临床药理学(12.3) ]。UGT1A1基因的遗传变异(例如UGT1A1 * 28等位基因)导致UGT1A1酶活性降低。对于UGT1A1 * 28等位基因纯合的个体,TRODELVY 引起中性粒细胞减少的风险增加[参见警告和注意事项(5.5)]。UGT1A1 * 28等位基因的纯合子约占黑人或非裔美国人人口的20%,白人人口的10%和东亚人口的2%。在某些人群中可能存在除UGT1A1 * 28外的功能等位基因减少。
13毒理学
13.1致癌,诱变,生育力受损
尚未对sacituzumab govitecan-hziy进行致癌性研究。
SN-38 在中国仓鼠卵巢细胞的体外哺乳动物细胞微核试验中具有致死性,在体外细菌反向突变(Ames)分析中不具有致突变性。
还没有进行过sacituzumab govitecan-hziy的生育力研究。在食蟹猴的重复剂量毒性研究中,剂量≥60 mg的第1天和第4天静脉注射sacituzumab govitecan-hziy导致子宫内膜萎缩,子宫出血,卵巢滤泡闭锁增加和阴道上皮细胞萎缩。 / kg(≥6倍于人体推荐剂量10 mg / kg,基于体重)。
14临床研究
的功效TRODELVY在研究IMMU-132-01(NCT01631552),多中心,单组,试验,登记108例谁收到了至少两个在先治疗转移性疾病转移性三阴性乳腺癌(mTNBC)评价。肿块大于7 cm的患者不符合条件。经过脑转移治疗的患者至少四周内未接受大剂量类固醇(> 20 mg泼尼松或同等剂量)。患有已知吉尔伯特病的患者被排除在外。
在21天的治疗周期的第1天和第8天,患者接受TRODELVY 10 mg / kg静脉滴注。对患者进行TRODELVY治疗,直到疾病进展或对该疗法不耐受为止。每8周进行一次肿瘤成像,并在最初的部分或完全缓解后4-6周获得确诊的CT / MRI扫描,直到需要治疗终止的进展。主要疗效结局指标是研究者使用RECIST 1.1和反应持续时间评估的总反应率(ORR)。
中位年龄为55岁(范围:31 – 80岁);87%的患者年龄小于65岁。大多数患者为女性(99%)和白人(76%)。在研究开始时,所有患者的ECOG表现状态均为0(29%)或1(71%)。内脏疾病占76%,肝转移为42%,肺/胸膜转移为56%,脑转移为2%。初诊时有12名患者(11%)患有IV期疾病。
在转移环境中接受的先前全身治疗的中位数为3(范围:2-10)。转移前的化学疗法包括卡铂或顺铂(69%),吉西他滨(55%),紫杉醇或多西他赛(53%),卡培他滨(51%),依立布林(45%),阿霉素(24%),长春瑞滨(16) %),环磷酰胺(19%)和ixabepilone(8%)。
总体而言,在(新)佐剂或转移性环境中,98%的患者曾接受过紫杉烷类药物,86%的患者曾接受过蒽环类药物。
表5总结了功效结果。
表5:IMMU-132-01中mTNBC患者的疗效结果

15参考
1.“ OSHA危险药物”。OSHA。http://www.osha.gov/SLTC/hazardousdrugs/index.html。
16供应/存储和处理方式
用于注射的TRODELVY(sacituzumab govitecan-hziy)是单剂量小瓶中的无菌,灰白色至淡黄色冻干粉末。每个TRODELVY小瓶都单独装在纸箱中:
- NDC 55135-132-01包含一个180 mg小瓶
将小瓶放在原始纸箱中的2°C至8°C(36°F至46°F)的冰箱中,以防光照,直到重新配制为止。不要冻结。
TRODELVY是一种细胞毒性药物。遵循适用的特殊处理和处置程序1。
17患者咨询信息
建议患者阅读FDA批准的患者标签(患者信息)
中性粒细胞减少症
告知患者中性粒细胞减少的风险。指示患者如果出现发烧,发冷或其他感染迹象,请立即与他们的医疗服务提供者联系[请参阅警告和注意事项(5.1)]。
腹泻
告知患者腹泻的风险。指导患者如果在治疗期间首次出现腹泻,应立即联系其医疗服务提供者;大便黑色或血腥;脱水症状,如头昏眼花,头晕或昏厥;由于恶心或呕吐而无法通过口摄入液体;或在24小时内无法控制腹泻[请参阅警告和注意事项(5.2)]。
过敏症
告知患者发生严重输注反应和过敏反应的风险。指导患者在以下过程中出现面部,嘴唇,舌头或喉咙肿胀,荨麻疹,呼吸困难,头晕,头晕,发冷,严厉,喘息,瘙痒,潮红,皮疹,低血压或发烧,立即联系他们的医疗保健提供者。输注后24小时内[请参阅警告和注意事项(5.3)]。
恶心,呕吐
告知患者恶心和呕吐的风险。还建议根据既定指南进行预防性用药,使用两种或三种药物治疗方案来预防化疗引起的恶心和呕吐(CINV)。临床上也可以采用其他止吐药,镇静剂和其他支持性措施。所有患者均应接受带回家的药物,以预防和治疗延迟的恶心和呕吐,并有明确的说明。指示患者如果感到恶心或呕吐,请立即联系其医疗保健提供者[请参阅警告和注意事项(5.4)]。
胚胎-胎儿毒性
建议女性患者如果怀孕或怀孕,请联系其医疗保健提供者。告知女性患者胎儿的风险和可能的怀孕损失[请参见“在特定人群中使用(8.1)”。
避孕
劝告有生殖潜力的女性患者在治疗期间和最后一次服用TRODELVY 后6个月内使用有效的避孕药[请参见在特定人群中使用(8.3)]。
建议具有生殖潜力的女性伴侣的男性患者在治疗期间和最后一次服用TRODELVY 后3个月内使用有效的避孕药[请参见在特定人群中使用(8.3)]。
哺乳期
劝告妇女在治疗期间以及最后一次服用TRODELVY 后1个月内不要进行母乳喂养[请参见在特定人群中使用(8.2)]。
不孕症
告知有生殖潜力的女性,TRODELVY可能会损害生育能力[请参阅在特定人群中使用(8.3)]。
由制造:
Immunomedics,Inc.300
The American Road
Morris Plains,NJ 07950,USA
美国牌照编号1737


主显示屏– 180 mg盒标签
NDC 55135-132-01
仅Rx
TRODELVY ™
赛妥珠单抗地精
注射用
每瓶180毫克
仅用于静脉输液
注意:细胞毒剂
使用前立即稀释并稀释单剂量小瓶
丢弃未使用的部分
1瓶

主显示屏– 180 mg瓶标签
NDC 55135-132-01
仅Rx
TRODELVY ™
赛妥珠单抗地精
注射用
每瓶180毫克
仅用于静脉输液
注意:细胞毒剂
单剂量小瓶
丢弃未使用的部分
1瓶

【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
本说明书来源于:美国FDA网站
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书。
WARNING: NEUTROPENIA AND DIARRHEA
- Severe neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis. Initiate anti-infective treatment in patient with febrile neutropenia without delay [see Warnings and Precautions (5.1)].
- Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. Administer atropine, if not contraindicated, for early diarrhea of any severity. At the onset of late diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide [see Warnings and Precautions (5.2)]. If severe diarrhea occurs, withhold TRODELVY until resolved to < Grade 1 and reduce subsequent doses [see Dosage and Administration (2.3)].
1 INDICATIONS AND USAGE
TRODELVY is indicated for the treatment of adult patients with metastatic triple-negative breast cancer (mTNBC) who have received at least two prior therapies for metastatic disease.
This indication is approved under accelerated approval based on tumor response rate and duration of response [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
2 DOSAGE AND ADMINISTRATION
2.1 Important Use Information
Do NOT substitute TRODELVY for or use with other drugs containing irinotecan or its active metabolite SN-38.
2.2 Recommended Dose and Schedule
The recommended dose of TRODELVY is 10 mg/kg administered as an intravenous infusion once weekly on Days 1 and 8 of 21-day treatment cycles. Continue treatment until disease progression or unacceptable toxicity. Do not administer TRODELVY at doses greater than 10 mg/kg.
Administer TRODELVY as an intravenous infusion only. Do not administer as an intravenous push or bolus.
First infusion: Administer infusion over 3 hours. Observe patients during the infusion and for at least 30 minutes following the initial dose, for signs or symptoms of infusion-related reactions [see Warning and Precautions (5.3)].
Subsequent infusions: Administer infusion over 1 to 2 hours if prior infusions were tolerated. Observe patients during the infusion and for at least 30 minutes after infusion.
Premedication
Prior to each dose of TRODELVY, premedication for prevention of infusion reactions and prevention of chemotherapy-induced nausea and vomiting (CINV) is recommended.
- Premedicate with antipyretics, H1 and H2 blockers prior to infusion, and corticosteroids may be used for patients who had prior infusion reactions.
- Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist, as well as other drugs as indicated).
2.3 Dose Modifications for Adverse Reactions
Infusion-related Reactions
Slow or interrupt the infusion rate of TRODELVY if the patient develops an infusion-related reaction. Permanently discontinue TRODELVY for life-threatening infusion-related reactions [see Warnings and Precautions (5.3)]
Dose Modifications for Adverse Reactions
Withhold or discontinue TRODELVY to manage adverse reactions as described in Table 1. Do not re-escalate the TRODELVY dose after a dose reduction for adverse reactions has been made.
Table 1: Dose Modifications for Adverse Reactions

2.4 Preparation for Administration
Reconstitution
- TRODELVY is a cytotoxic drug.
- Follow applicable special handling and disposal procedures1.
- Calculate the required dose (mg) of TRODELVY based on the patient's body weight at the beginning of each treatment cycle (or more frequently if the patient's body weight changed by more than 10% since the previous administration) [see Dosage and Administration (2.1)].
- Allow the required number of vials to warm to room temperature.
- Using a sterile syringe, slowly inject 20 mL of 0.9% Sodium Chloride Injection, USP, into each 180 mg TRODELVY vial. The resulting concentration will be 10 mg/mL.
- Gently swirl vials and allow to dissolve for up to 15 minutes. Do not shake. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The solution should be free of visible particulates, clear and yellow. Do not use the reconstituted solution if it is cloudy or discolored.
- Use immediately to prepare a diluted TRODELVY infusion solution.
Dilution
- Calculate the required volume of the reconstituted TRODELVY solution needed to obtain the appropriate dose according to patient's body weight. Withdraw this amount from the vial(s) using a syringe. Discard any unused portion remaining in the vial(s).
- Slowly inject the required volume of reconstituted TRODELVY solution into a polypropylene (PP) infusion bag, to minimize foaming. Do not shake the contents.
- Adjust the volume in the infusion bag as needed with 0.9% Sodium Chloride Injection, USP, to obtain a concentration of 1.1 mg/mL to 3.4 mg/mL (total volume should not exceed 500 mL). For patients whose body weight exceeds 170 kg, divide the total dosage of TRODELVY equally between two 500 mL infusion bags and infuse sequentially via slow infusion.
- Only 0.9% Sodium Chloride Injection, USP, should be used since the stability of the reconstituted product has not been determined with other infusion-based solutions. Use the diluted solution in the infusion bag immediately. If not used immediately, the infusion bag containing TRODELVY solution can be stored refrigerated 2°C to 8°C (36°F to 46°F) for up to 4 hours. After refrigeration, administer diluted solution within 4 hours (including infusion time).
Do Not Freeze or Shake. Protect from Light.
Administration
- Administer TRODELVY as an intravenous infusion. Protect infusion bag from light.
- An infusion pump may be used.
- Do not mix TRODELVY, or administer as an infusion, with other medicinal products.
- Upon completion of the infusion, flush the intravenous line with 20 mL 0.9% Sodium Chloride Injection, USP.
3 DOSAGE FORMS AND STRENGTHS
For injection: 180 mg off-white to yellowish lyophilized powder in a single-dose vial.
4 CONTRAINDICATIONS
TRODELVY is contraindicated in patients who have experienced a severe hypersensitivity reaction to TRODELVY [see Warnings and Precautions (5.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Neutropenia
TRODELVY can cause severe or life-threatening neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 on Day 1 of any cycle or neutrophil count below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Dose modifications may be required due to neutropenia [see Dosage and Administration (2.3)].
Febrile neutropenia occurred in 6% (24/408) patients treated with TRODELVY, including 8% (9/108) patients with mTNBC after at least two prior therapies. Less than 1% (1/408) of patients had febrile neutropenia leading to permanent discontinuation.
The incidence of Grade 1-4 neutropenia was 64% in patients with mTNBC (n=108). In all patients treated with TRODELVY (n=408), the incidence of Grade1-4 neutropenia was 54%; Grade 4 neutropenia occurred in 13%. Less than 1% (2/408) of patients permanently discontinued treatment due to neutropenia.
5.2 Diarrhea
TRODELVY can cause severe diarrhea. Withhold TRODELVY for Grade 3-4 diarrhea at the time of scheduled treatment administration and resume when resolved to ≤ Grade 1 [see Dosage and Administration (2.3)].
At the onset of diarrhea, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated.
Patients who exhibit an excessive cholinergic response to treatment with TRODELVY (e.g., abdominal cramping, diarrhea, salivation, etc.) can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Diarrhea occurred in 63% (68/108) of patients with mTNBC and 62% (254/408) of all patients treated with TRODELVY. In each population, events of Grade 3-4 occurred in 9% (10/108) of mTNBC patients and 9% (36/408) of all patients treated with TRODELVY. Four out of 408 patients (<1%) discontinued treatment because of diarrhea. Neutropenic colitis was observed in 2% (2/108) of patients in the mTNBC cohort and 1% of all patients treated with TRODELVY.
5.3 Hypersensitivity
TRODELVY can cause severe and life-threatening hypersensitivity. Anaphylactic reactions have been observed in clinical trials with TRODELVY.
Hypersensitivity reactions within 24 hours of dosing occurred in 37% (151/408) of patient treated with TRODELVY. Grade 3-4 hypersensitivity occurred in 1% (6/408) of patients treated with TRODELVY. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 1% (3/408).
Pre-infusion medication for patients receiving TRODELVY is recommended. Observe patients closely for infusion-related reactions during each TRODELVY infusion and for at least 30 minutes after completion of each infusion [see Dosage and Administration (2.3)]. Medication to treat such reactions, as well as emergency equipment, should be available for immediate use.
5.4 Nausea and Vomiting
TRODELVY is emetogenic. Nausea occurred in 69% (74/108) of patients with mTNBC and 69% (281/408) of all patients treated with TRODELVY. Grade 3 nausea occurred in 6% (7/108) and 5% (22/408) of these populations, respectively.
Vomiting occurred in 49% (53/108) of patients with mTNBC and 45% (183/408) of all patients treated with TRODELVY. Grade 3 vomiting occurred in 6% (7/108) and 4% (16/408) of these patients, respectively.
Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK-1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV).
Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting at the time of scheduled treatment administration and resume with additional supportive measures when resolved to Grade ≤ 1 [see Dosage and Administration (2.3)].
Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
5.5 Use in Patients with Reduced UGT1A1 Activity
Individuals who are homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia and may be at increased risk for other adverse reactions following initiation of TRODELVY treatment.
In 84% (343/408) of patients who received TRODELVY (up to 10 mg/kg on Days 1 and 8 of a 21-day cycle) and had retrospective UGT1A1 genotype results available, the incidence of Grade 4 neutropenia was 26% (10/39) in patients homozygous for the UGT1A1*28 allele, 13% (20/155) in patients heterozygous for the UGT1A1*28 allele and 11% (16/149) in patients homozygous for the wild-type allele [see Clinical Pharmacology (12.5)].
Closely monitor patients with reduced UGT1A1 activity for severe neutropenia. The appropriate dose for patients who are homozygous for UGT1A1*28 is not known and should be considered based on individual patient tolerance to treatment [see Dosage and Administration (2.3)].
5.6 Embryo-Fetal Toxicity
Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1)]. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Neutropenia [see Warnings and Precautions (5.1)]
- Diarrhea [see Warnings and Precautions (5.2)]
- Hypersensitivity [see Warnings and Precautions (5.3)]
- Nausea and Vomiting [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The data described in the Warnings and Precautions section reflect exposure to TRODELVY as a single agent in a single-arm, open-label study (IMMU-132-01) in 408 patients with mTNBC and other malignancies who had received prior systemic therapeutic regimen for advanced disease. TRODELVY was administered as an intravenous infusion once weekly on Days 1 and 8 of 21-day treatment cycles at doses up to 10 mg/kg until disease progression or unacceptable toxicity.
The data in Table 2 reflect exposure to TRODELVY in a subset of 108 patients with mTNBC who had received at least two prior treatments for metastatic disease in study (IMMU-132-01). Patients received TRODELVY 10 mg/kg via intravenous infusion on Days 1 and 8 of 21-day treatment cycles until disease progression or unacceptable toxicity. The median treatment duration in these 108 patients was 5.1 months (range: 0-51 months).
Serious adverse reactions were reported in 31% of the patients. The most frequent serious adverse reactions (reported in >1%) of the patients receiving TRODELVY were febrile neutropenia (6%) vomiting (5%), nausea (3%), dyspnea (3%), diarrhea (4%), anemia (2%), pleural effusion, neutropenia, pneumonia, dehydration (each 2%).
TRODELVY was permanently discontinued for adverse reactions in 2% of patients. Adverse reactions leading to discontinuation were anaphylaxis, anorexia/fatigue, headache (each <1%, 1 patient for each event). Forty- five percent (45%) of patients experienced an adverse reaction leading to treatment interruption. The most common adverse reaction leading to treatment interruption was neutropenia (33%). Adverse reactions leading to dose reduction occurred in 33% of patients treated with TRODELVY, with 24% having one dose reduction and 9% with two dose reductions. The most common adverse reaction leading to dose reductions was neutropenia/febrile neutropenia.
Adverse reactions occurring in ≥10% of patients with mTNBC in the IMMU-132-01 study are summarized in Table 2.
Table 2: Adverse Reactions in ≥ 10% of Patients with mTNBC in IMMU-132-01


Table 3: Laboratory Abnormalities observed in >10% of Patients while receiving TRODELVY

6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other sacituzumab govitecan products may be misleading.
The analysis of immunogenicity of TRODELVY in serum samples from 106 patients with mTNBC was evaluated using an electrochemiluminescence (ECL)-based immunoassay to test for anti-sacituzumab govitecan-hziy antibodies. Detection of the anti-sacituzumab govitecan-hziy antibodies was done using a 3-tier approach: screen, confirm, and titer. Persistent anti-sacituzumab govitecan-hziy antibodies developed in 2% (2/106) of patients.
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on TRODELVY
UGT1A1 Inhibitors
Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38 [see Warning and Precaution (5.5) and Clinical Pharmacology (12.3, 12.5)]. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers
Exposure to SN-38 may be substantially reduced in patients concomitantly receiving UGT1A1 enzyme inducers [see Warning and Precaution (5.5) and Clinical Pharmacology (12.3, 12.5)]. Avoid administering UGT1A1 inducers with TRODELVY.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. There are no available data in pregnant women to inform the drug-associated risk. TRODELVY contains a genotoxic component, SN-38, and is toxic to rapidly dividing cells [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1)]. Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 – 4% and 15 – 20%, respectively.
Data
Animal data
There were no reproductive and developmental toxicology studies conducted with sacituzumab govitecan-hziy.
8.2 Lactation
Risk Summary
There is no information regarding the presence of sacituzumab govitecan-hziy or SN-38 in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment and for 1 month after the last dose of TRODELVY.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to the initiation of TRODELVY.
Contraception
Females
TRODELVY can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose.
Males
Because of the potential for genotoxicity, advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
Infertility
Females
Based on findings in animals, TRODELVY may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of TRODELVY have not been established in pediatric patients.
8.5 Geriatric Use
Of the patients who received TRODELVY, 19/108 (18%) patients with mTNBC and 144/408 (35%) of all patients were ≥ 65 years old. No overall differences in safety and effectiveness were observed between these patients and younger patients.
8.6 Hepatic Impairment
No adjustment to the starting dose is required when administering TRODELVY to patients with mild hepatic impairment (bilirubin less than or equal to 1.5 ULN and AST/ALT < 3 ULN).
The exposure of TRODELVY in patients with mild hepatic impairment (bilirubin less than or equal to ULN and AST greater than ULN, or bilirubin greater than 1.0 to 1.5 ULN and AST of any level; n=12) was similar to patients with normal hepatic function (bilirubin or AST less than ULN; n=45).
The safety of TRODELVY in patients with moderate or severe hepatic impairment has not been established. TRODELVY has not been tested in patients with serum bilirubin > 1.5 ULN, or AST and ALT > 3 ULN, or AST and ALT > 5 ULN and associated with liver metastases.
No dedicated trial was performed to investigate the tolerability of TRODELVY in patients with moderate or severe hepatic impairment. No recommendations can be made for the starting dose in these patients.
10 OVERDOSAGE
In a clinical trial, planned doses of up to 18 mg/kg (approximately 1.8 times the maximum recommended dose of 10 mg/kg) of TRODELVY were administered. In these patients, a higher incidence of severe neutropenia was observed.
11 DESCRIPTION
Sacituzumab govitecan-hziy is a Trop-2 directed antibody and topoisomerase inhibitor conjugate, composed of the following three components:
- the humanized monoclonal antibody, hRS7 IgG1ϰ (also called sacituzumab), which binds to Trop-2 (the trophoblast cell-surface antigen-2);
- the drug SN-38, a topoisomerase inhibitor;
- a hydrolysable linker (called CL2A), which links the humanized monoclonal antibody to SN-38.
The recombinant monoclonal antibody is produced by mammalian (murine myeloma) cells, while the small molecule components SN-38 and CL2A are produced by chemical synthesis. Sacituzumab govitecan-hziy contains on average 7 to 8 molecules of SN-38 per antibody molecule. Sacituzumab govitecan-hziy has a molecular weight of approximately 160 kilodaltons. Sacituzumab govitecan-hziy has the following chemical structure.
TRODELVY (sacituzumab govitecan-hziy) for injection is a sterile, preservative-free, off-white to yellowish lyophilized powder for intravenous use in a 50 mL clear glass single-dose vial, with a rubber stopper and crimp-sealed with an aluminum flip-off cap.
Each single-dose vial of TRODELVY delivers 180 mg sacituzumab govitecan-hziy, 77.3 mg 2-(N-morpholino) ethane sulfonic acid (MES), 1.8 mg polysorbate 80 and 154 mg trehalose dihydrate. Reconstitution with 20 mL of 0.9% Sodium Chloride Injection, USP, results in a concentration of 10 mg/mL with a pH of 6.5.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sacituzumab govitecan-hziy is a Trop-2-directed antibody-drug conjugate. Sacituzumab is a humanized antibody that recognizes Trop-2. The small molecule, SN-38, is a topoisomerase I inhibitor, which is covalently attached to the antibody by a linker. Pharmacology data suggest that sacituzumab govitecan-hziy binds to Trop-2-expressing cancer cells and is internalized with the subsequent release of SN-38 via hydrolysis of the linker. SN-38 interacts with topoisomerase I and prevents re-ligation of topoisomerase I-induced single strand breaks. The resulting DNA damage leads to apoptosis and cell death. Sacituzumab govitecan-hziy decreased tumor growth in mouse xenograft models of triple-negative breast cancer.
12.2 Pharmacodynamics
Exposure-response relationships and the time course of pharmacodynamics response are unknown for sacituzumab govitecan-hziy.
12.3 Pharmacokinetics
The serum pharmacokinetics of sacituzumab govitecan-hziy and SN-38 were evaluated in a study in a population of mTNBC patients who received sacituzumab govitecan-hziy as a single agent at a dose of 10 mg/kg. The pharmacokinetic parameters of sacituzumab govitecan-hziy and free SN-38 are presented in Table 4.
Table 4: Summary of Mean PK Parameters (±Standard Deviation) of Sacituzumab Govitecan-hziy and Free SN-38

Distribution
The mean volume of distribution for sacituzumab govitecan-hziy was 0.045 L/kg.
Elimination
The mean half-life of sacituzumab govitecan-hziy and free SN-38 was 16 and 18 hours, respectively. The clearance of the sacituzumab govitecan-hziy was 0.002 L/h/kg.
Metabolism
No metabolism studies with sacituzumab govitecan-hziy have been conducted. SN-38 (the small molecule moiety of sacituzumab govitecan-hziy) is metabolized via UGT1A1. The glucuronide metabolite of SN-38 (SN-38G) was detectable in the serum of patients.
Specific Populations
Pharmacokinetic analyses in a limited number of patients with mTNBC (n = 57) did not identify an effect of age or race on the pharmacokinetics of sacituzumab govitecan-hziy. Renal elimination is known to contribute minimally to the excretion of SN-38, the small molecule moiety of sacituzumab govitecan-hziy. There are no data on the pharmacokinetics of sacituzumab govitecan-hziy in patients with renal impairment or end-stage renal disease (CLcr ≤ 30 mL/min).
The exposure of sacituzumab govitecan-hziy is similar in patients with mild hepatic impairment (bilirubin less than or equal to ULN and AST greater than ULN, or bilirubin greater than 1.0 to less than 1.5 ULN and AST of any level; n=12) to patients with normal hepatic function (bilirubin or AST less than ULN; n=45).
Sacituzumab govitecan-hziy exposure is unknown in patients with moderate or severe hepatic impairment. SN-38 exposure may be elevated in such patients due to decreased hepatic UGT1A1 activity.
Drug Interaction Studies
No drug-drug interaction studies were conducted with sacituzumab govitecan-hziy or its components Inhibitors or inducers of UGT1A1 are expected to increase or decrease SN-38 exposure, respectively [see Drug Interactions (7)].
12.5 Pharmacogenomics
SN-38 is metabolized via UGT1A1 [see Clinical Pharmacology (12.3)]. Genetic variants of the UGT1A1 gene such as the UGT1A1*28 allele lead to reduced UGT1A1 enzyme activity. Individuals who are homozygous for the UGT1A1*28 allele are at increased risk for neutropenia from TRODELVY [see Warnings and Precautions (5.5)]. Approximately 20% of the Black or African American population, 10% of the White population, and 2% of the East Asian population are homozygous for the UGT1A1*28 allele. Decreased function alleles other than UGT1A1*28 may be present in certain populations.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with sacituzumab govitecan-hziy.
SN-38 was clastogenic in an in vitro mammalian cell micronucleus test in Chinese hamster ovary cells and was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay.
Fertility studies with sacituzumab govitecan-hziy have not been conducted. In a repeat-dose toxicity study in cynomolgus monkeys, intravenous administration of sacituzumab govitecan-hziy on Day 1 and Day 4 resulted in endometrial atrophy, uterine hemorrhage, increased follicular atresia of the ovary, and atrophy of vaginal epithelial cells at doses ≥ 60 mg/kg (≥ 6 times the human recommended dose of 10 mg/kg based on body weight).
14 CLINICAL STUDIES
The efficacy of TRODELVY was evaluated in study IMMU-132-01 (NCT01631552), a multicenter, single-arm, trial that enrolled 108 patients with metastatic triple-negative breast cancer (mTNBC) who had received at least two prior treatments for metastatic disease. Patients with bulky disease, defined as a mass >7 cm, were not eligible. Patients with treated brain metastases not receiving high dose steroids (>20 mg prednisone or equivalent) for at least four weeks were eligible. Patients with known Gilbert's disease were excluded.
Patients received TRODELVY 10 mg/kg intravenously on Days 1 and 8 of a 21-day treatment cycle. Patients were treated with TRODELVY until disease progression or intolerance to the therapy. Tumor imaging was obtained every 8 weeks, with confirmatory CT/MRI scans obtained 4-6 weeks after an initial partial or complete response, until progression requiring treatment discontinuation. Major efficacy outcome measures were investigator assessed overall response rate (ORR) using RECIST 1.1 and duration of response.
The median age was 55 years (range: 31 – 80 years); 87% of patients were younger than 65 years. The majority of patients were female (99%), and White (76%). At study entry, all patients had an ECOG performance status of 0 (29%) or 1 (71%). Seventy-six percent had visceral disease, 42% had hepatic metastases, 56% had lung/pleura metastases, and 2% had brain metastases. Twelve patients (11%) had Stage IV disease at the time of initial diagnosis.
The median number of prior systemic therapies received in the metastatic setting was 3 (range: 2 - 10). Prior chemotherapies in the metastatic setting included carboplatin or cisplatin (69%), gemcitabine (55%), paclitaxel or docetaxel (53%), capecitabine (51%), eribulin (45%), doxorubicin (24%), vinorelbine (16%), cyclophosphamide (19%), and ixabepilone (8%).
Overall, 98% of patients had received prior taxanes and 86% had received prior anthracyclines either in the (neo)adjuvant or metastatic setting.
Table 5 summarizes the efficacy results.
Table 5: Efficacy results for patients with mTNBC in IMMU-132-01

15 REFERENCES
1. “OSHA Hazardous Drugs.” OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
16 HOW SUPPLIED/STORAGE AND HANDLING
TRODELVY (sacituzumab govitecan-hziy) for injection is a sterile, off-white to yellowish lyophilized powder in a single-dose vial. Each TRODELVY vial is individually boxed in a carton:
- NDC 55135-132-01 contains one 180 mg vial
Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until time of reconstitution. Do not freeze.
TRODELVY is a cytotoxic drug. Follow applicable special handling and disposal procedures1.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information)
Neutropenia
Advise patients of the risk of neutropenia. Instruct patients to immediately contact their healthcare provider if they experience fever, chills, or other signs of infection [see Warnings and Precautions (5.1)].
Diarrhea
Advise patients of the risk of diarrhea. Instruct patients to immediately contact their healthcare provider if they experience diarrhea for the first time during treatment; black or bloody stools; symptoms of dehydration such as lightheadedness, dizziness, or faintness; inability to take fluids by mouth due to nausea or vomiting; or inability to get diarrhea under control within 24 hours [see Warnings and Precautions (5.2)].
Hypersensitivity
Inform patients of the risk of serious infusion reactions and anaphylaxis. Instruct patients to immediately contact their healthcare provider if they experience facial, lip, tongue, or throat swelling, urticaria, difficulty breathing, lightheadedness, dizziness, chills, rigors, wheezing, pruritus, flushing, rash, hypotension or fever, that occur during or within 24 hours following the infusion [see Warnings and Precautions (5.3)].
Nausea/Vomiting
Advise patients of the risk of nausea and vomiting. Premedication according to established guidelines with a two or three drug regimen for prevention of chemotherapy-induced nausea and vomiting (CINV) is also recommended. Additional antiemetics, sedatives, and other supportive measures may also be employed as clinically indicated. All patients should receive take-home medications for preventing and treating delayed nausea and vomiting, with clear instructions. Instruct patients to immediately contact their healthcare provider if they experience uncontrolled nausea or vomiting [see Warnings and Precautions (5.4)].
Embryo-Fetal Toxicity
Advise female patients to contact their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1)].
Contraception
Advise female patients of reproductive potential to use effective contraception during treatment and for 6 months after the last dose of TRODELVY [see Use in Specific Populations (8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of TRODELVY [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment and for 1 month after the last dose of TRODELVY [see Use in Specific Populations (8.2)].
Infertility
Advise females of reproductive potential that TRODELVY may impair fertility [see Use in Specific Populations (8.3)].
Manufactured by:
Immunomedics, Inc.
300 The American Road
Morris Plains, NJ 07950, USA
U.S. License No. 1737


Principal Display Panel – 180 mg Box Label
NDC 55135-132-01
Rx only
TRODELVY™
sacituzumab
govitecan-hziyFor Injection
180 mg per vial
For intravenous infusion only
CAUTION: Cytotoxic Agent
Reconstitute and dilute immediately
prior to useSingle-dose vial
Discard unused portion
1 vial

Principal Display Panel – 180 mg Vial Label
NDC 55135-132-01
Rx only
TRODELVY™
sacituzumab
govitecan-hziyFor Injection
180 mg per vial
For intravenous infusion only
CAUTION: Cytotoxic Agent
Single-dose vial
Discard unused portion
1 vial

【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。