商品详情
返回产品目录

商品包装及说明书因厂家更换频繁,如有不符以实物为主
注射用戈沙妥珠单抗
国际零售参考价:¥**/瓶
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。

1 适应症和用法
1.1 局部晚期或转移性乳腺癌
- TRODELVY适用于治疗患有不可切除的局部晚期或转移性三阴性乳腺癌( mTNBC )的成年患者,这些患者已接受过两种或多种先前的全身治疗,其中至少一种是针对转移性疾病。
- TRODELVY适用于治疗患有不可切除的局部晚期或转移性激素受体 (HR) 阳性、人表皮生长因子受体 2 (HER2) 阴性(IHC 0、IHC 1+ 或 IHC 2+/ISH–)乳腺癌的成年患者在转移环境中接受过内分泌治疗和至少两种额外全身治疗的癌症患者。
1.2 局部晚期或转移性尿路上皮癌
- TRODELVY适用于治疗先前接受过含铂化疗和程序性死亡受体 1 (PD-1) 或程序性死亡配体 1 (PD-L1) 的局部晚期或转移性尿路上皮癌 (mUC) 成年患者)抑制剂。
该适应症根据肿瘤反应率和反应持续时间在加速批准下获得批准[见临床研究 (14.3) ]。对该适应症的持续批准可能取决于确认试验中对临床益处的验证和描述。
2 剂量和给药方法
2.1 重要使用信息
不要用TRODELVY代替或与含有伊立替康或其活性代谢物 SN-38 的其他药物一起使用。
2.2 推荐剂量
TRODELVY的推荐剂量为 10 mg/kg,在 21 天治疗周期的第 1 天和第 8 天每周静脉输注一次。继续治疗直至疾病进展或不可接受的毒性。不要以大于 10 mg/kg 的剂量施用TRODELVY 。
仅将TRODELVY作为静脉输注给药。不要作为静脉推注或推注给药。
首次输注:输注超过 3 小时。输注期间和初始剂量后共至少 30 分钟观察患者输注相关反应的体征或症状[见警告和注意事项(5.3) ]。
随后的输注:如果之前的输注是耐受的,则输注超过 1 至 2 小时。输注期间和输注后至少 30 分钟观察患者。
术前用药
在每次服用TRODELVY之前,建议预先用药以预防输液反应和化疗引起的恶心和呕吐( CINV )。
- 输注前用退热药、H1 和 H2 阻滞剂预先给药,并且皮质类固醇可用于先前有输注反应的患者。
- 使用两种或三种药物组合方案进行预先用药(例如,地塞米松与 5-HT3 受体拮抗剂或 NK 1受体拮抗剂,以及指定的其他药物)。
2.3 对不良反应剂量修改
输液相关反应
如果患者出现输液相关反应,请减慢或中断TRODELVY的输液速度。因危及生命的输液相关反应而永久停用TRODELVY [见警告和注意事项 (5.3) ]
不良反应的剂量修改
如表 1 所述,停止或停止TRODELVY以处理不良反应。在针对不良反应减少剂量后,不要重新增加TRODELVY剂量。
表 1:针对不良反应的剂量修改
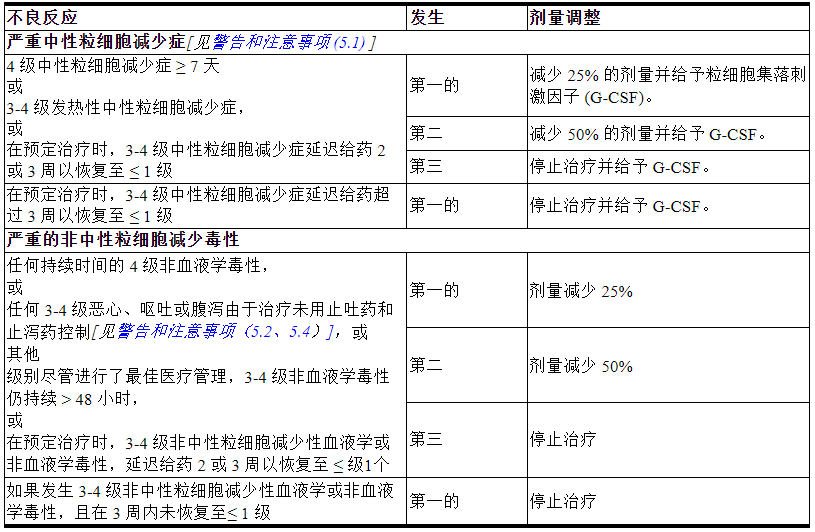
2.4 制备和给药
重构
- TRODELVY是一种危险药物。
- 遵循适用的特殊处理和处置程序1。
- 根据每个治疗周期开始时患者的体重计算 TRODELVY 的所需剂量(mg)(或者如果自上次给药以来患者的体重变化超过 10%,则更频繁)[见剂量和给药方法( 2.2 ) ]。
- 让所需数量的小瓶升温至室温。
- 使用无菌注射器,将 20 mL 0.9% 氯化钠注射液 (USP) 缓慢注入每个 180 mg TRODELVY小瓶中。每个小瓶都装有溢出物,以补偿制备过程中的液体损失,并在重构后产生的总体积提供10 mg/mL 的浓度。
- 轻轻旋转小瓶并使其溶解长达 15 分钟。不要摇晃。只要溶液和容器允许,肠胃外药物产品在给药前应目视检查是否有颗粒物质和变色。该溶液应不含可见颗粒,呈透明和黄色。如果混浊或变色,请勿使用重新配制的溶液。
- 立即使用以制备稀释的TRODELVY输液。
稀释
- 根据患者的体重计算获得适当剂量所需的重组TRODELVY溶液的所需量。
- 确定输液溶液的最终体积,以在 1.1 mg/mL 至 3.4 mg/mL 的TRODELVY浓度范围内提供适当的剂量。
- 仅使用 0.9% 氯化钠注射液,USP,因为重组TRODELVY溶液的稳定性尚未用其他输液溶液确定。使用聚氯乙烯、聚丙烯/聚乙烯、聚烯烃或乙烯醋酸乙烯酯输液袋。
- 在添加计算量的重组 TRODELVY溶液后,从最终输液袋中取出并丢弃达到指定TRODELVY浓度所需的 0.9% 氯化钠注射液 USP 体积。
- 使用注射器从小瓶中取出计算量的重构TRODELVY溶液。丢弃小瓶中剩余的任何未使用部分。
- 为尽量减少泡沫,将计算量的重构TRODELVY溶液缓慢注入输液袋。不要摇动内容物。
- 如果不立即使用,含有TRODELVY溶液的输液袋可以在 2°C 至 8°C(36°F 至 46°F)的温度下避光保存长达 24 小时。冷藏后,在 8 小时内(包括输注时间)在室温至 25°C (77°F) 下给予稀释溶液。
不要冷冻或摇晃。
行政
- 将TRODELVY作为静脉输注给药。输液袋避光。在给患者给药期间应盖好输液袋,直至给药完成。输液过程中无需遮盖输液管或使用避光管。
- 可以使用输液泵。
- 不要将TRODELVY与其他医药产品混合或作为输液给药。
- 输注完成后,用 20 mL 0.9% 氯化钠注射液 (USP) 冲洗静脉内管路。
3 剂型和强度
注射用:180 mg 灰白色至淡黄色冻干粉,装在单剂量小瓶中。
4 禁忌症
TRODELVY禁用于对TRODELVY 有过严重超敏反应的患者[见警告和注意事项 (5.3) ]。
5 警告和注意事项
5.1 中性粒细胞减少
接受TRODELVY治疗的患者可能会出现严重、危及生命或致命的中性粒细胞减少症。64% 接受TRODELVY治疗的患者出现中性粒细胞减少症。49% 的患者出现 3-4 级中性粒细胞减少症。6% 的患者出现发热性中性粒细胞减少症。首次发作中性粒细胞减少症(包括发热性中性粒细胞减少症)的中位时间为 16 天并且在一些患者群体中发生得更早[见警告和注意事项(5.5) ]。1.4% 的患者发生中性粒细胞减少性结肠炎。
如果在任何周期的第 1 天中性粒细胞绝对计数低于 1500/mm 3或在任何周期的第 8 天中性粒细胞计数低于 1000/mm 3 ,则停止使用TRODELVY。停止使用TRODELVY治疗中性粒细胞减少性发热。由于中性粒细胞减少,可能需要调整剂量。如临床指示或表 1 中指示给予 G-CSF [见剂量和给药方法 (2.3) ]。
5.2 腹泻
TRODELVY可引起严重腹泻。所有接受TRODELVY治疗的患者中有 64% 发生腹泻。所有接受TRODELVY治疗的患者中有 11% 发生 3-4 级腹泻。一名患者在腹泻后出现肠穿孔。0.7% 的患者发生腹泻导致脱水和随后的急性肾损伤。
在预定的治疗给药时停用TRODELVY治疗 3-4 级腹泻,并在解决至≤1 级时恢复[见剂量和给药方法 (2.3) ]。
腹泻发作时,评估感染原因,如果阴性,立即开始使用洛哌丁胺,初始剂量为 4 mg,随后每次腹泻发作时剂量为 2 mg,每日最大剂量为 16 mg。腹泻消退后 12 小时停用洛哌丁胺。也可根据临床指征采用额外的支持措施(例如,补液和电解质)。
对TRODELVY治疗表现出过度胆碱能反应(例如,腹部绞痛、腹泻、流涎等)的患者可以接受适当的术前用药(例如,阿托品)以进行后续治疗。
5.3 超敏性和输注相关反应
TRODELVY治疗已发生严重的超敏反应,包括危及生命的过敏反应。严重体征和症状包括心脏骤停、低血压、喘息、血管性水肿、肿胀、肺炎和皮肤反应[见禁忌症 (4) ]。
35% 接受TRODELVY治疗的患者在给药后 24 小时内发生超敏反应。2% 接受TRODELVY治疗的患者发生 3-4 级超敏反应。导致永久停用TRODELVY的超敏反应发生率为 0.2%。过敏反应的发生率为0.2%。
建议对接受TRODELVY的患者进行输液反应的术前用药。有药物和应急设备来治疗输注相关反应,包括过敏反应,在给予TRODELVY 时可立即使用[见剂量和给药方法 (2.2) ]。
在每次TRODELVY输注期间和每次输注完成后至少 30 分钟密切监测患者的超敏反应和输注相关反应[见剂量和给药方法 (2.3) ]。
因 4 级输液相关反应永久停用TRODELVY [见剂量和给药方法 (2.3) ]。
5.4 恶心和呕吐
TRODELVY会致吐。所有接受TRODELVY治疗的患者中有 64% 出现恶心。3% 的患者发生 3-4 级恶心。
所有接受TRODELVY治疗的患者中有 35% 发生呕吐。这些患者中有 2% 发生了 3-4 级呕吐。
使用两种或三种药物联合方案(例如,地塞米松与 5-HT3 受体拮抗剂或 NK 1受体拮抗剂以及其他药物)进行预给药,以预防化疗引起的恶心和呕吐(CINV)[见剂量和管理(2.2) ]。
在计划的治疗给药时停止3 级恶心或 3-4 级呕吐的TRODELVY剂量,并在解决 ≤ 1 级时用额外的支持措施恢复[见剂量和给药方法 (2.3) ]。
还可以根据临床指示使用额外的止吐药和其他支持措施。应为所有患者提供带回家的药物,并附有预防和治疗恶心和呕吐的明确说明。
5.5 在有减低 UGT1A1 活性患者不良反应风险增加
尿苷二磷酸-葡萄糖醛酸转移酶 1A1 (UGT1A1)*28 等位基因纯合的患者患中性粒细胞减少症、发热性中性粒细胞减少症和贫血的风险增加;并且在使用TRODELVY治疗时可能会增加其他不良反应的风险。
在 948 名接受TRODELVY且具有 UGT1A1 基因型结果的患者中分析了中性粒细胞减少症和贫血的发生率。在 UGT1A1 *28 等位基因纯合的患者 (n=112) 中,3-4 级中性粒细胞减少症的发生率为 58%。在 UGT1A1*28 等位基因杂合的患者 (n=420) 中,3-4 级中性粒细胞减少症的发生率为 49%。在对野生型等位基因纯合的患者(n=416)中,3-4级中性粒细胞减少的发生率为43% [见临床药理学(12.5) ]。在 UGT1A1 *28 等位基因纯合的患者中,3-4 级贫血的发生率为 21%。在 UGT1A1*28 等位基因杂合的患者中,3-4 级贫血的发生率为 10%。在野生型等位基因纯合的患者中,3-4 级贫血的发生率为 9%。
UGT1A1*28 等位基因纯合子患者首次出现中性粒细胞减少症(包括发热性中性粒细胞减少症)的中位时间为 9 天,UGT1A1*28 等位基因杂合子患者为 15 天,野生型等位基因纯合子患者为 20 天。UGT1A1*28 等位基因纯合子患者首次贫血的中位时间为 21 天,UGT1A1*28 等位基因杂合子患者为 25 天,野生型等位基因纯合子患者为 28 天。
密切监测已知 UGT1A1 活性降低的患者的不良反应。根据对有急性早发或异常严重不良反应证据的患者观察到的不良反应的发作、持续时间和严重程度的临床评估,暂停或永久停用 TRODELVY,这可能表明 UGT1A1 酶活性降低[见剂量和给药方法(2.3 ) ) ]。
5.6 胚胎-胎儿毒性
根据其作用机制,TRODELVY在给孕妇服用时会导致致畸和/或胚胎-胎儿致死。TRODELVY含有一种基因毒性成分 SN-38,并靶向快速分裂的细胞[参见临床药理学 (12.1)和非临床毒理学 (13.1) ]。忠告孕妇和有生殖潜能的女性对胎儿的潜在风险。建议具有生殖潜力的女性在TRODELVY治疗期间和最后一次给药后 6 个月内使用有效的避孕措施。建议有生殖潜力女性伴侣的男性患者在TRODELVY治疗期间使用有效的避孕措施和最后一次给药后 3 个月[见在特定人群中使用 (8.1 , 8.3) ]。
6 不良反应
标签的其他部分更详细地讨论了以下不良反应:
- 中性粒细胞减少症[见警告和注意事项 (5.1) ]
- 腹泻[见警告和注意事项 (5.2) ]
- 超敏反应和输液相关反应[见警告和注意事项 (5.3) ]
- 恶心和呕吐[见警告和注意事项 (5.4) ]
6.1 临床试验经验
因为临床试验是在广泛不同条件下进行的,一种药物临床试验中观察到的不良反应率不能与另一种药物临床试验中的发生率直接比较并且可能不反映临床实践中观察到的发生率。
警告和注意事项部分描述的汇总安全人群反映了来自四项研究 IMMU-132-01、ASCENT、TROPiCS-02 和 TROPHY 的 1063 名患者接触 TRODELVY 的情况,其中包括 366 名 mTNBC 患者、322 名激素受体阳性患者(HR+)/人表皮生长因子受体 2 阴性 (HER2-) 乳腺癌和 180 名 mUC 患者。TRODELVY在 21 天治疗周期的第 1 天和第 8 天每周一次静脉输注,剂量为 10 mg/kg,直至疾病进展或出现不可接受的毒性。在接受TRODELVY治疗的 1063 名患者中,中位治疗持续时间为 4.1 个月(范围:0 至 63 个月)。在这个汇集的安全人群中,包括实验室异常在内的最常见(≥ 25%)的不良反应是白细胞计数减少(84%)、中性粒细胞计数减少(75%)、血红蛋白减少(69%)、腹泻(64%)、恶心(64%)、淋巴细胞计数减少 (63%)、疲劳 (51%)、脱发 (45%)、便秘 (37%)、葡萄糖增加 (37%)、白蛋白减少 (35%)、呕吐 (35%) 、食欲下降 (30%)、肌酐清除率下降 (28%)、碱性磷酸酶上升 (28%)、镁下降 (27%)、钾下降 (26%) 和钠下降 (26%)。
局部晚期或转移性三阴性乳腺癌
上升研究
TRODELVY的安全性在一项随机、主动对照、开放标签研究(ASCENT)中进行了评估,研究对象为先前接受过紫杉烷类药物和至少两种既往化疗的 mTNBC 患者。患者被随机化(1:1)接受TRODELVY(n=258)或单药化疗(n=224),并接受治疗直至疾病进展或不可接受的毒性[见临床研究(14.1) ]。对于接受TRODELVY治疗的患者,中位治疗持续时间为 4.4 个月(范围:0 至 23 个月)。
27% 接受TRODELVY的患者发生严重不良反应。接受TRODELVY治疗的患者中 > 1% 的严重不良反应包括中性粒细胞减少症(7%)、腹泻(4%)和肺炎(3%)。接受TRODELVY治疗的患者中有 1.2% 发生致命不良反应,包括呼吸衰竭(0.8%)和肺炎(0.4%)。 5% 的患者因不良反应而永久停用TRODELVY 。≥ 1 % 接受TRODELVY的患者导致永久停药的不良反应是肺炎(1%)和疲劳(1%)。
63% 的患者发生了导致TRODELVY治疗中断的不良反应。导致治疗中断的最常见(≥5%)不良反应是中性粒细胞减少症(47%)、腹泻(5%)、呼吸道感染(5%)和白细胞减少症(5%)。
22% 的患者发生了导致TRODELVY剂量减少的不良反应。导致剂量减少的最常见 (>4%) 不良反应是中性粒细胞减少 (11%) 和腹泻 (5%)。
44%接受TRODELVY治疗的患者使用了粒细胞集落刺激因子(G-CSF)。
表 2 和 3 分别总结了 ASCENT 研究中的不良反应和实验室异常。
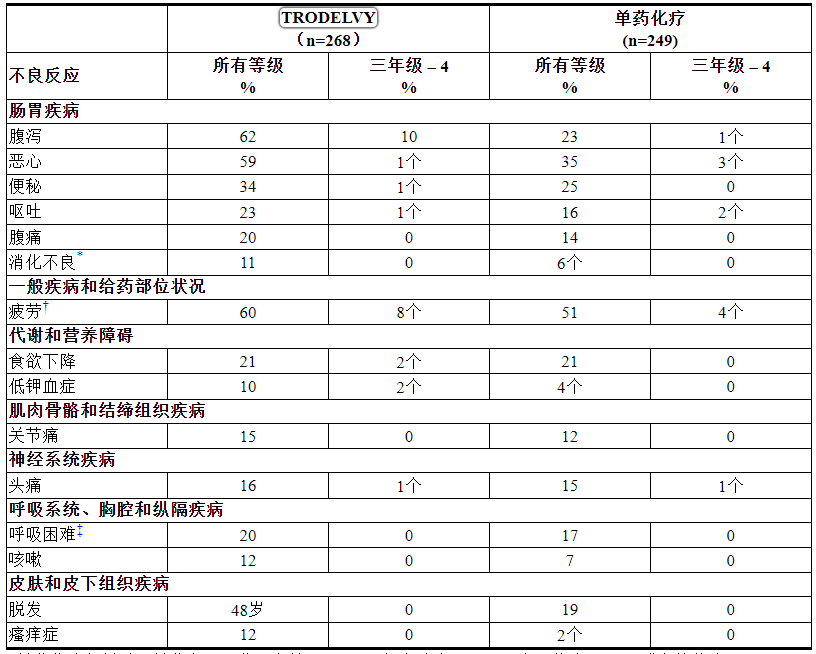
表 2:在 ASCENT 中 ≥ 10% 的 mTNBC 患者的不良反应
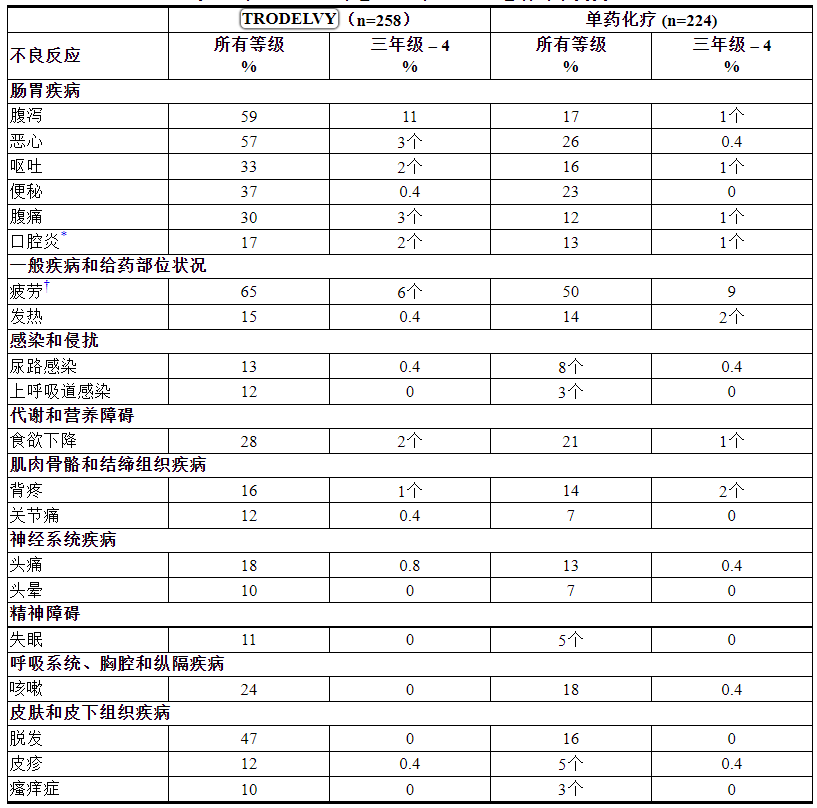
*单药化疗包括以下单药之一:艾日布林 (n=139)、卡培他滨 (n=33)、吉西他滨 (n=38) 或长春瑞滨(除非患者有 ≥ 2 级神经病变,n=52) .
根据 NCI CTCAE v.5.0 分级。
*包括口腔炎、舌炎、口腔溃疡、粘膜炎症
†包括疲劳和乏力
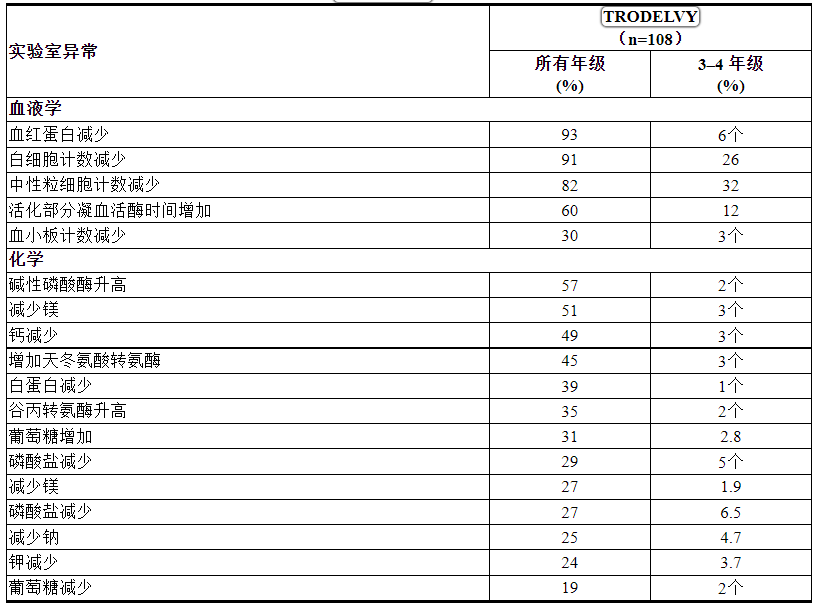
表 3:在 ASCENT 中 > 10% 的 mTNBC 患者存在实验室异常
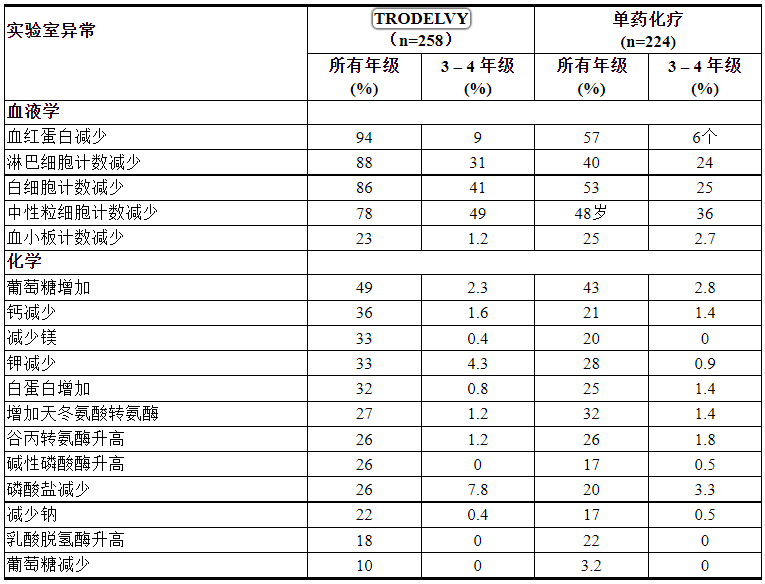
研究 IMMU-132-01
TRODELVY的安全性在一项针对 mTNBC 和其他恶性肿瘤患者的单臂、开放标签研究 (IMMU-132-01) 中进行了评估,该研究包括 108 名 mTNBC 患者,他们之前接受过至少两种针对转移性疾病的抗癌治疗[参见临床研究 (14.1) ]。 TRODELVY在 21 天治疗周期的第 1 天和第 8 天每周一次静脉输注,剂量高达 10 mg/kg,直至疾病进展或出现不可接受的毒性。这 108 名患者的中位治疗持续时间为 5.1 个月(范围:0 至 51 个月)。
31%的患者发生严重不良反应。接受TRODELVY治疗的患者中 > 1% 的严重不良反应包括发热性中性粒细胞减少症 (6%) 呕吐 (5%)、恶心 (3%)、呼吸困难 (3%)、腹泻 (4%)、贫血 (2%)、胸腔积液、粒细胞减少、肺炎、脱水(各2%)。
2% 的患者因不良反应而永久停用TRODELVY 。导致永久停药的不良反应为过敏反应、厌食/疲劳、头痛(各 0.9%)。百分之四十五 (45%) 的患者经历了导致治疗中断的不良反应。导致治疗中断的最常见不良反应是中性粒细胞减少症 (33%)。33% 接受TRODELVY治疗的患者发生了导致剂量减少的不良反应,其中 24% 减少了一次剂量,9% 减少了两次剂量。导致剂量减少的最常见不良反应是中性粒细胞减少症/发热性中性粒细胞减少症。
表 4 和表 5 总结了 IMMU-132-01 研究中≥10% 的 mTNBC 患者发生的不良反应和实验室异常。
表 4:IMMU-132-01 中 ≥ 10% 的 mTNBC 患者的不良反应
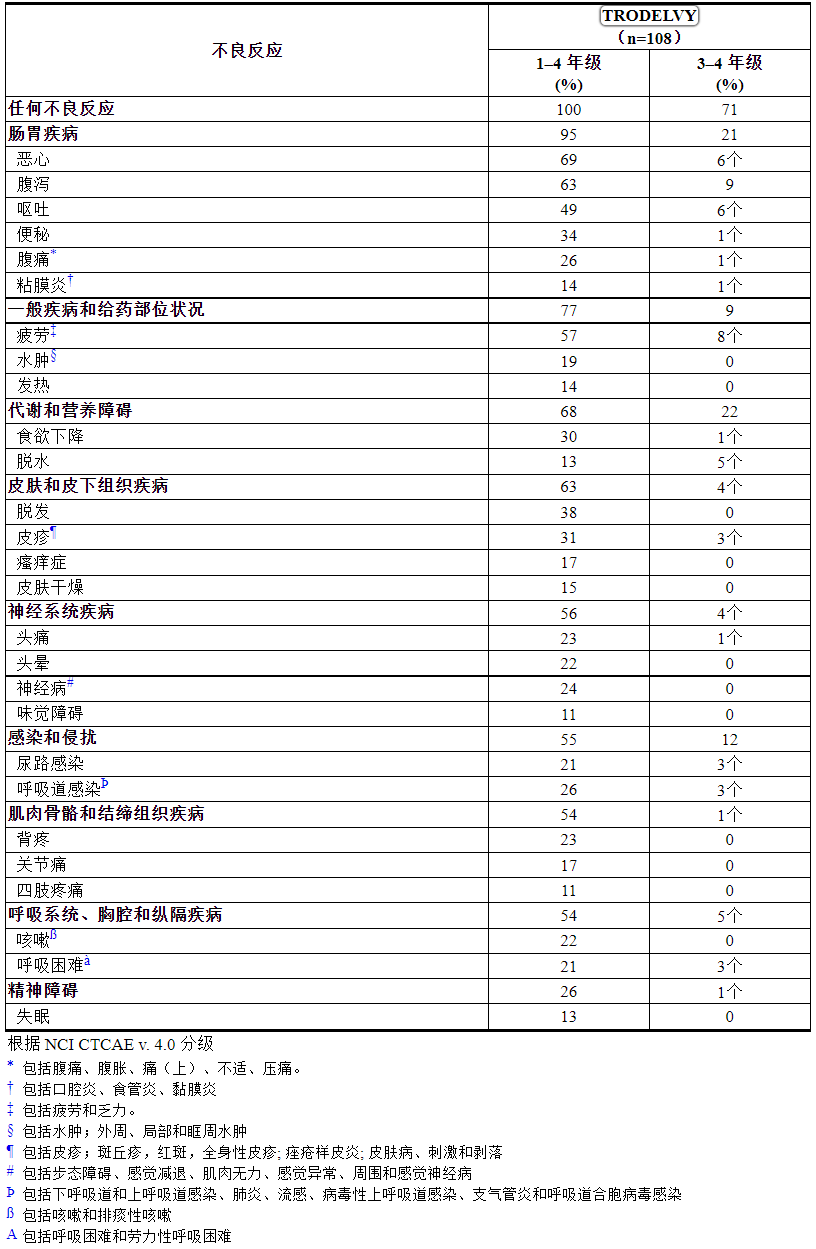
表 5:在 IMMU-132-01 中接受TRODELVY时观察到≥ 10% 的患者的实验室异常
局部晚期或转移性 HR 阳性、HER2 阴性乳腺癌
TROPiCS-02 研究
TRODELVY的安全性在一项随机、主动控制、开放标签研究 (TROPiCS-02) 中进行了评估,该研究的对象是无法切除的局部晚期或转移性 HR 阳性、HER2 阴性乳腺癌患者,其疾病在以下任何一种治疗后出现进展背景:CDK 4/6 抑制剂、内分泌治疗和紫杉烷;患者在转移性环境中至少接受过两种先前的化疗(如果在 12 个月内发生进展,其中一种可以在新辅助或辅助环境中进行)。患者被随机化(1:1)接受TRODELVY(n=268)或单药化疗(n=249)并接受治疗直至疾病进展或不可接受的毒性[见临床研究(14.2) ]。TRODELVY的中位治疗持续时间为 4.1 个月(范围:0 至 63 个月)。
28% 接受TRODELVY的患者发生严重不良反应。接受TRODELVY治疗的患者中 >1% 的严重不良反应包括腹泻(5%)、发热性中性粒细胞减少症(4%)、中性粒细胞减少症(3%)、腹痛、结肠炎、中性粒细胞减少性结肠炎、肺炎和呕吐(各 2%)。2% 的接受TRODELVY治疗的患者发生了致命的不良反应,包括心律失常、COVID-19、神经系统疾病、肺栓塞和感染性休克(各 0.4%)。6% 的患者因不良反应而永久停用TRODELVY 。导致接受TRODELVY治疗的患者永久停药的最常见 (≥0.5%) 不良反应是虚弱、一般身体健康状况恶化和中性粒细胞减少症(各 0.7%)。
66% 的患者发生了导致TRODELVY治疗中断的不良反应。导致治疗中断的最常见(≥5%)不良反应是中性粒细胞减少症(50%)。
33% 的患者发生了导致TRODELVY剂量减少的不良反应。导致剂量减少的最常见 (>5%) 不良反应是中性粒细胞减少 (16%) 和腹泻 (8%)。54% 接受TRODELVY的患者使用了 G-CSF 。
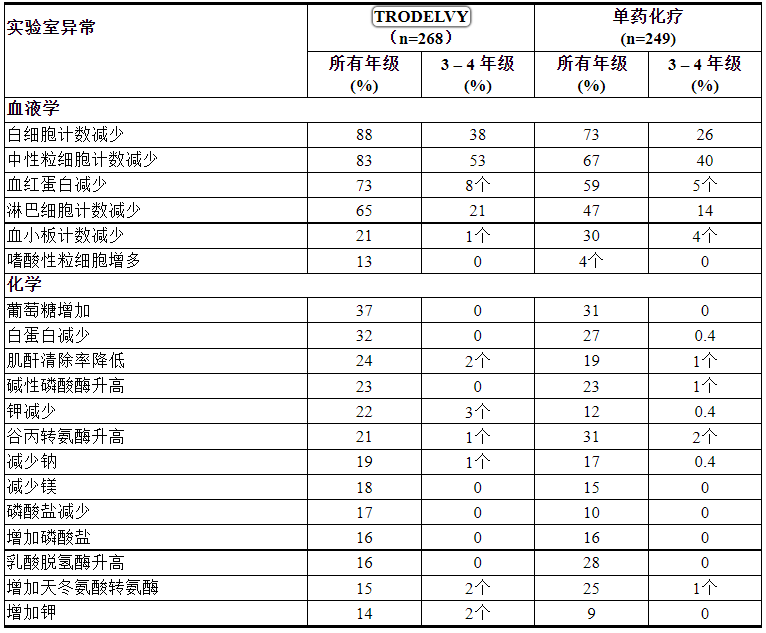
表 6 和 7 总结了 TROPiCS-02 研究中的不良反应和实验室异常。
表 6:TROPiCS-02 中 ≥ 10% HR+/HER2- mBC 患者的不良反应
*单药化疗包括以下单药之一:艾日布林 (n=130)、长春瑞滨 (n=63)、吉西他滨 (n=56) 或卡培他滨 (n=22)。
根据 NCI CTCAE v.5.0 分级。
*包括消化不良、胃食管反流病。
†包括疲劳、乏力。
‡包括呼吸困难;劳力性呼吸困难
TROPiCS-02 的其他临床显着不良反应(≤ 10%)包括:低血压(5%)、疼痛(5%)、流鼻涕(5%)、低钙血症(3%)、鼻塞(3%)、皮肤色素沉着过度、 (3%)、结肠炎或中性粒细胞减少性结肠炎 (2%)、低钠血症 (2%)、肺炎 (2%)、蛋白尿 (1%)、肠炎 (0.4%)。
表 7:TROPiCS-02 中 > 10% 的 HR+/HER2- mBC 患者的实验室异常
局部晚期或转移性尿路上皮癌
奖杯研究
TRODELVY的安全性在一项单臂、开放标签研究中进行了评估,研究对象为 mUC 患者(n=113),这些患者既往接受过铂类和抗 PD-1/PD-L1 治疗。 TRODELVY在 21 天治疗周期的第 1 天和第 8 天每周一次静脉输注,剂量为 10 mg/kg,直至疾病进展或出现不可接受的毒性。(见临床研究 14.3)
44%的患者发生严重不良反应。接受TRODELVY治疗的患者中 >1% 的严重不良反应包括感染 (18%)、中性粒细胞减少症 (12%,包括 10% 的发热性中性粒细胞减少症)、急性肾损伤 (6%)、尿路感染 (6%)、败血症或菌血症(5%)、腹泻 (4%)、贫血、静脉血栓栓塞和小肠梗阻(各 3%)、肺炎、腹痛、发热和血小板减少症(各 2%)。3.6%的患者发生致命不良反应,包括败血症、呼吸衰竭、鼻出血和自杀。
10% 的患者因不良反应而永久停用TRODELVY 。导致永久停用研究药物的最常见不良反应是中性粒细胞减少症(4%,包括 2% 的发热性中性粒细胞减少症)。52% 的患者发生了导致剂量中断的不良反应。导致剂量中断的最常见不良反应是中性粒细胞减少症(27%,包括 2% 的发热性中性粒细胞减少症)、感染(12%)和急性肾损伤(8%)。导致TRODELVY剂量减少的不良反应发生在 42% 的患者中。导致剂量减少的最常见(> 4%)不良反应是中性粒细胞减少(13%,包括 3% 的发热性中性粒细胞减少)、腹泻(11%)、疲劳(8%)和感染(4%)。47%接受TRODELVY治疗的患者使用了粒细胞集落刺激因子(G-CSF)。
包括实验室异常在内的最常见(≥ 25%)不良反应为白细胞计数减少(78%)、腹泻(72%)、血红蛋白减少(71%)、淋巴细胞计数减少(71%)、中性粒细胞计数减少(67%) ,恶心(66%),葡萄糖增加(59%),疲劳(56%),白蛋白减少(51%),脱发(49%),钙减少(46%),钠减少(43%),食欲下降( 41%)、磷酸盐减少 (41%)、碱性磷酸酶增加 (36%)、便秘 (34%)、呕吐 (34%)、活化部分凝血活酶时间增加 (33%)、肌酐增加 (32%)、镁减少(31%)、谷丙转氨酶升高 (28%)、乳酸脱氢酶升高 (28%)、腹痛 (27%)、钾减少 (27%)、天冬氨酸转氨酶升高 (26%) 和血小板计数降低 (25%) ).
表 8 和 9 总结了 TROPHY 研究中≥10% 的 mUC 患者发生的不良反应和实验室异常。
表 8: TROPHY中 ≥ 15%(1-4 级)或 ≥ 5%(≥ 3 级)接受TRODELVY治疗的患者报告的不良反应
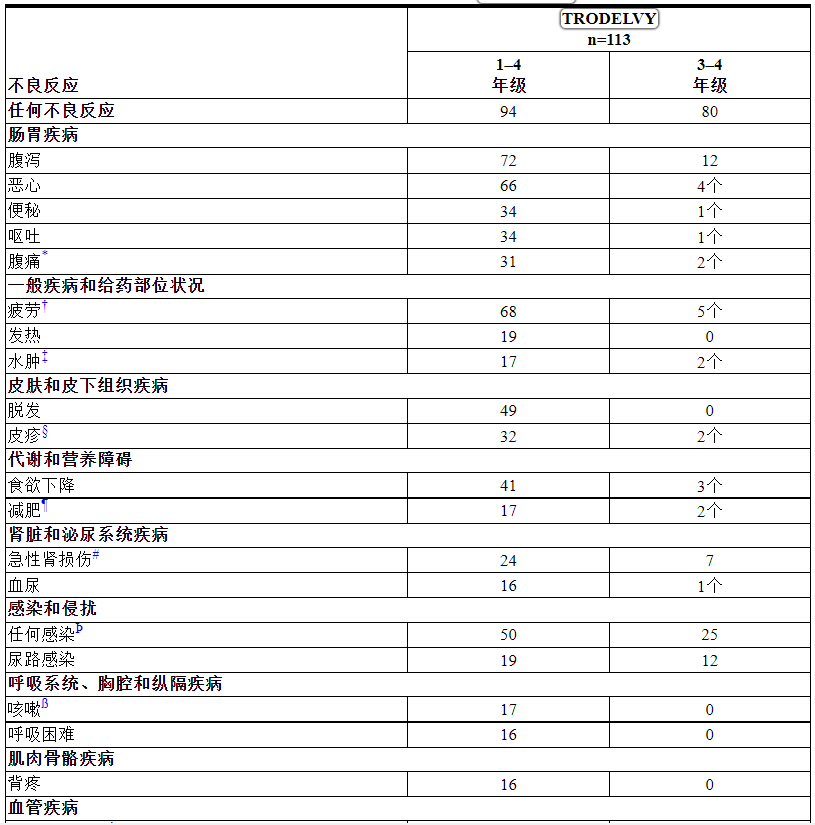
根据 NCI CTCAE v.5.0 分级。
*包括腹部不适、腹痛、下腹痛、上腹痛、胃肠痛
†包括疲劳和乏力
‡包括生殖器水肿、外周水肿、外周肿胀
§包括痤疮样皮炎、大疱性皮炎、红斑、扁平苔藓、光敏反应、瘙痒症、全身性瘙痒症、皮疹、斑疹、斑丘疹、瘙痒性皮疹、皮肤乳头状瘤、皮肤毒性
¶包括发育迟缓和体重下降
#包括急性肾损伤、血肌酐升高、肾病毒性、肾功能衰竭、肾功能损害
Þ包括菌血症、体癣、支气管炎、念珠菌感染、蜂窝组织炎、艰难梭菌感染、冠状病毒感染、器械相关感染、憩室炎、大肠杆菌菌血症、大肠杆菌肾盂肾炎、毛囊炎、肠胃炎、大肠杆菌肠胃炎、带状疱疹、肾脏感染、克雷伯菌败血症、肺部感染、鼻咽炎、口腔念珠菌病、口腔疱疹、肺炎、肾盂肾炎、急性肾盂肾炎、呼吸道感染、鼻炎、败血症、鼻窦炎、皮肤感染、牙脓肿、上呼吸道感染、尿路感染、尿脓毒症、血管装置感染、病毒感染、病毒性咽炎、外阴阴道霉菌感染
ß包括咳嗽、咳痰、上呼吸道咳嗽综合征
A包括深静脉血栓形成、栓塞和肺栓塞
TROPHY 中其他有临床意义的不良反应(≤15%)包括:周围神经病变(12%)、败血症或菌血症(9%)和肺炎(4%)。
表 9: TROPHY中 ≥ 20%(任何级别)或 ≥ 5%(3-4 级)接受TRODELVY治疗的患者报告的实验室异常
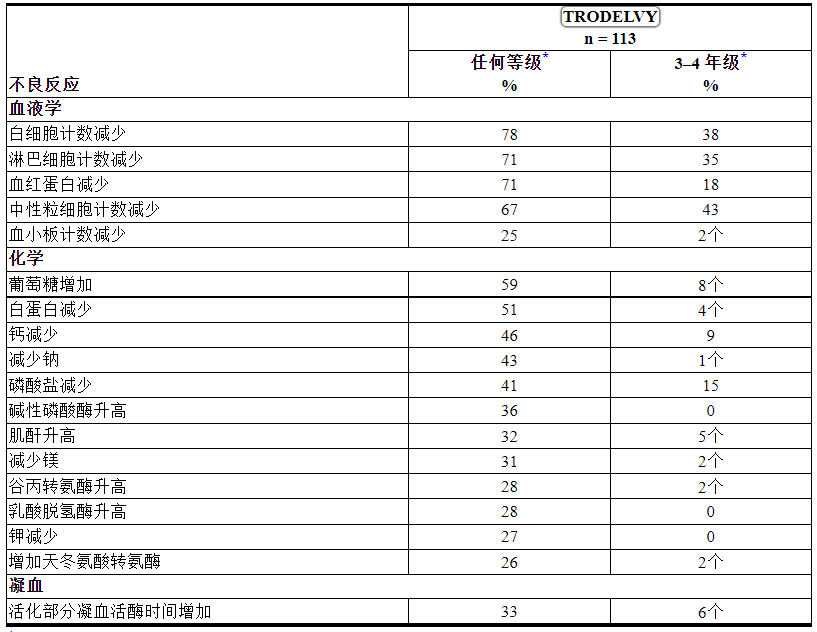
*每个实验室参数的分母基于具有可用基线和治疗后实验室值的患者数量(范围:66 至 111 名患者)。
7 药物相互作用
7.1 其他药物对TRODELVY的影响
UGT1A1抑制剂
TRODELVY与 UGT1A1 抑制剂的同时给药可能会增加不良反应的发生率,因为可能会增加对 SN-38 的全身暴露[见警告和注意事项(5.5)和临床药理学( 12.3,12.5 ) ]。避免将 UGT1A1 抑制剂与TRODELVY一起使用。
UGT1A1 诱导剂
在同时接受 UGT1A1 酶诱导剂的患者中可能减少对 SN-38 的暴露[见警告和注意事项 (5.5)和临床药理学 (12.3 , 12.5) ]。避免将 UGT1A1 诱导剂与TRODELVY一起使用。
8 在特定人群中的使用
8.1 怀孕
风险总结
根据其作用机制,TRODELVY在给孕妇服用时会导致致畸和/或胚胎-胎儿致死。在孕妇中没有可用的数据来告知与药物相关的风险。TRODELVY含有遗传毒性成分 SN-38,对快速分裂的细胞有毒性[见临床药理学 (12.1)和非临床毒理学 (13.1) ]。忠告孕妇和有生殖潜能的女性对胎儿的潜在风险。
对指定人群的主要先天缺陷和流产的估计背景风险未知。在美国一般人群中,临床认可的妊娠中重大出生缺陷和流产的估计背景风险分别为 2 – 4% 和 15 – 20%。
数据
动物数据
没有对 sacituzumab govitecan-hziy 进行生殖和发育毒理学研究。
8.2 哺乳期
风险总结
没有关于母乳中 sacituzumab govitecan-hziy 或 SN-38 的存在、对母乳喂养儿童的影响或对产奶量的影响的信息。由于母乳喂养的孩子可能会出现严重的不良反应,建议女性在治疗期间和最后一剂 TRODELVY 后 1 个月内不要母乳喂养。
8.3 具有生殖潜能的女性和男性
妊娠试验
在开始TRODELVY之前验证具有生殖潜力的女性的怀孕状态。
避孕
女性
给孕妇服用TRODELVY会造成胎儿伤害[见在特定人群中的使用(8.1) ]。建议具有生殖潜力的女性在TRODELVY治疗期间和最后一次给药后 6 个月内使用有效的避孕措施。
男性
由于潜在的遗传毒性,建议有生殖潜力女性伴侣的男性患者在TRODELVY治疗期间和最后一次给药后 3 个月内使用有效的避孕措施。
不育症
女性
根据在动物身上的发现,TRODELVY可能会损害具有生殖潜能的女性的生育能力[见非临床毒理学 (13.1) ]。
8.4 儿科使用
尚未在儿科患者中确定TRODELVY的安全性和有效性。
8.5 老年人使用
在接受TRODELVY治疗的 366 名 TNBC 患者中,19% 的患者年龄为 65 岁,3% 的患者年龄在 75 岁及以上。在≥ 65 岁的患者和较年轻的患者之间未观察到安全性和有效性的总体差异。
在接受TRODELVY治疗的 322 名 HR+/HER2- 乳腺癌患者中,26% 的患者年龄在 65 岁及以上,6% 的患者年龄在 75 岁及以上。在≥ 65 岁的患者和较年轻的患者之间未观察到有效性的总体差异。与年轻患者 (3%) 相比,65 岁或以上患者 (14%) 因不良反应而停药的比例更高。
在接受TRODELVY治疗的 180 名 UC 患者中,59% 的患者年龄在 65 岁及以上,27% 的患者年龄在 75 岁及以上。在≥ 65 岁的患者和较年轻的患者之间未观察到有效性的总体差异。与年轻患者 (8%) 相比,65 岁或以上患者 (14%) 因不良反应而停药的比例更高。
8.6 肝损伤
轻度肝功能不全患者服用TRODELVY时无需调整起始剂量[见临床药理学 (12.3) ]。
尚未确定 TRODELVY 在中度(总胆红素 > 1.5 至 3.0 × ULN)或重度(总胆红素 > 3.0 × 正常上限 [ULN])肝功能损害患者中的安全性。TRODELVY尚未在 AST 或 ALT > 3 ULN 且无肝转移,或 AST 或 ALT > 5 ULN 且有肝转移的患者中进行过测试。不能对这些患者的起始剂量提出建议。
10 过量
在一项临床试验中,给予的TRODELVY计划剂量高达 18 mg/kg(约为最大推荐剂量 10 mg/kg 的 1.8 倍) 。在这些患者中,观察到严重中性粒细胞减少症的发生率较高。
11 描述
Sacituzumab govitecan-hziy 是一种 Trop-2 定向抗体和拓扑异构酶抑制剂偶联物,由以下三个组分组成:
- 人源化单克隆抗体 hRS7 IgG1κ(也称为 sacituzumab),它与 Trop-2(滋养层细胞表面抗原 2)结合;
- 药物 SN-38,一种拓扑异构酶抑制剂;
- 一个可水解的接头(称为 CL2A),它将人源化单克隆抗体连接到 SN-38。
重组单克隆抗体由哺乳动物(鼠类骨髓瘤)细胞产生,而小分子成分SN-38和CL2A由化学合成产生。Sacituzumab govitecan-hziy 每个抗体分子平均含有 7 至 8 个 SN-38 分子。Sacituzumab govitecan-hziy 的分子量约为 160 道尔顿。Sacituzumab govitecan-hziy 具有以下化学结构。
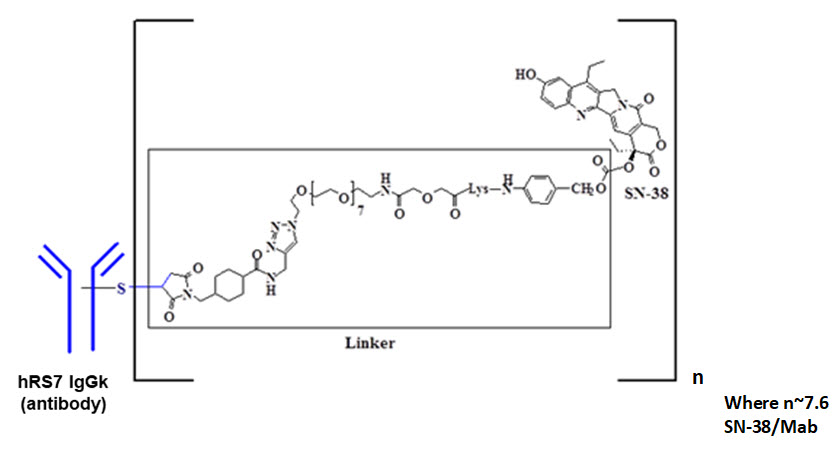
注射用TRODELVY (sacituzumab govitecan-hziy) 是一种无菌、不含防腐剂、灰白色至淡黄色冻干粉,用于静脉内使用,装在 50 mL 透明玻璃单剂量小瓶中,带有橡胶塞,并用铝盖卷曲密封-关闭上限。
TRODELVY的每个单剂量小瓶可提供 180 mg sacituzumab govitecan-hziy、77.3 mg 2-(N-吗啉基)乙磺酸 (MES)、1.8 mg 聚山梨酯 80 和 154 mg 海藻糖二水合物。用 20 mL 的 0.9% 氯化钠注射液 (USP) 重构,浓度为 10 mg/mL,pH 值为 6.5。
12 临床药理学
12.1 作用机制
Sacituzumab govitecan-hziy 是一种针对 Trop-2 的抗体-药物偶联物。Sacituzumab 是一种识别 Trop-2 的人源化抗体。小分子 SN-38 是一种拓扑异构酶 I 抑制剂,它通过接头共价连接到抗体上。药理学数据表明,sacituzumab govitecan-hziy 与表达 Trop-2 的癌细胞结合,并随着随后通过接头水解释放 SN-38 而被内化。SN-38 与拓扑异构酶 I 相互作用并防止拓扑异构酶 I 诱导的单链断裂重新连接。由此产生的 DNA 损伤导致细胞凋亡和细胞死亡。Sacituzumab govitecan-hziy 可降低小鼠三阴性乳腺癌异种移植模型中的肿瘤生长。
12.2 药效学
TRODELVY暴露-反应关系和疗效的药效学时程尚未得到充分表征。
心脏电生理学
在推荐剂量下,相对于基线的最大平均变化为 9.7 毫秒(双侧 90% 置信区间的上限为 16.8 毫秒)。在 QTc 增加和 SN-38 浓度之间观察到正暴露-反应关系。
12.3 药代动力学
在 mBC 患者中评估了 sacituzumab govitecan-hziy 和 SN-38 的血清药代动力学,这些患者接受了 10 mg/kg 剂量的 sacituzumab govitecan-hziy 作为单一药物。表 10 中列出了 sacituzumab govitecan-hziy 和游离 SN-38 的药代动力学参数。
表 10:Sacituzumab Govitecan-hziy 和游离 SN-38 的平均 PK 参数 (CV%) 汇总*
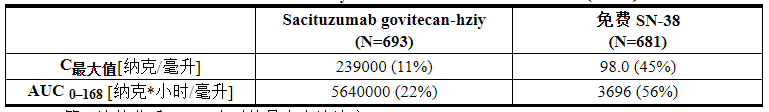
C max:第一次给药后 0-168 小时的最大血清浓度
AUC 0–168:首次给药后 168 小时血清浓度曲线下面积
*基于群体 PK 分析估计的参数
分配
根据群体药代动力学分析,sacituzumab govetican-hziy 的稳态分布容积为 3.6L。
消除
在转移性三阴性乳腺癌患者中,sacituzumab govitecan-hziy 和游离 SN-38 的中位消除半衰期 (t 1/2 ) 分别为 23.4 和 17.6 小时。根据群体药代动力学分析,sacituzumab govitecan-hziy 的估计平均清除率 (%CV) 为 0.13 L/h (12%)。
代谢
尚未对 sacituzumab govitecan-hziy 进行代谢研究。SN-38(sacituzumab govitecan-hziy 的小分子部分)通过 UGT1A1 代谢。在患者的血清中可检测到 SN-38 的葡萄糖醛酸代谢物 (SN-38G)。
特定人群
接受TRODELVY治疗的患者的药代动力学分析未发现年龄(27 至 88 岁)、种族(白人、黑人或亚裔)或轻度肾功能损害至中度肾功能损害(CLcr 30 至 89 mL/min)对sacituzumab govitecan-hziy 的药代动力学。已知肾脏消除对 SN-38 的排泄贡献最小,SN-38 是 sacituzumab govitecan-hziy 的小分子部分。没有关于 sacituzumab govitecan-hziy 在严重肾功能不全(CLcr 15 至 29 mL/min)或终末期肾病(CLcr < 15 mL/min)患者中的药代动力学数据。
肝功能不全患者
sacituzumab govitecan-hziy 在轻度肝功能不全患者(总胆红素 ≤ ULN 伴 AST > ULN,或胆红素 >1.0 至 ≤ 1.5 ULN 伴任何 AST;n=257)与肝功能正常患者(总胆红素或 AST < ULN;n=526)。
在有中度(总胆红素 > 1.5 至 3.0 × ULN)或严重(总胆红素 > 3.0 × ULN)肝受损患者中,Sacituzumab govitecan-hziy 和游离 SN-38 暴露未知。
药物相互作用研究
未对 sacituzumab govitecan-hziy 或其成分进行药物相互作用研究。UGT1A1 的抑制剂或诱导剂可能分别增加或减少 SN-38 暴露[见药物相互作用 (7) ]。
12.5 药物基因组学
SN-38 通过 UGT1A1 代谢 [见临床药理学 (12.3) ]。UGT1A1 基因的遗传变异,例如 UGT1A1*28 等位基因,会导致 UGT1A1 酶活性降低。与野生型个体(* 1/*1) 相比,UGT1A1*28 等位基因纯合或杂合的个体患中性粒细胞减少症、发热性中性粒细胞减少症和贫血的风险增加 (*1/* 1) [见警告和注意事项 (5.5) ]。大约 20% 的黑人或非裔美国人人口、10% 的白人人口和 2% 的东亚人口是 UGT1A1*28 等位基因 (*28/*28) 的纯合子。大约 40% 的黑人或非裔美国人人口、50% 的白人人口和 25% 的东亚人口是 UGT1A1*28 等位基因杂合子 (*1/*28)。UGT1A1*28 以外的功能降低的等位基因可能存在于某些人群中。
12.6 免疫原性
抗体形成的检测高度依赖于检测的灵敏度和特异性。测定方法的差异排除了将下述研究中的抗药抗体发生率与其他研究(包括 TRODELVY 的研究)中的抗药抗体发生率进行有意义的比较。
在接受TRODELVY治疗的患者的临床研究中,在中位 4 个月的治疗期间,785 名患者中有 9 名(1.1%)产生了针对 sacituzumab govitecan 的抗体;这些患者中有 6 名(占接受TRODELVY治疗的所有患者的 0.8% )具有针对 sacituzumab govitecan 的中和抗体。由于抗药抗体的发生率较低,这些抗体对 sacituzumab govitecan 的药代动力学、药效学、安全性和/或有效性的影响尚不清楚。
13 非临床毒理学
13.1 癌发生、突变发生、生育力受损
尚未用 sacituzumab govitecan-hziy 进行致癌性研究。
SN-38 在中国仓鼠卵巢细胞的体外哺乳动物细胞微核试验中具有致突变性,在体外细菌回复突变 (Ames) 试验中没有致突变性。
尚未进行 sacituzumab govitecan-hziy 的生育力研究。在食蟹猴重复给药毒性研究中,第 1 天和第 4 天静脉内给予 sacituzumab govitecan-hziy 剂量≥ 60 mg 导致子宫内膜萎缩、子宫出血、卵巢卵泡闭锁增加和阴道上皮细胞萎缩/kg(≥ 6 倍人类推荐剂量 10 mg/kg,基于体重)。
14 项临床研究
14.1 局部晚期或转移性三阴性乳腺癌
上升
一项多中心、开放标签、随机研究(ASCENT;NCT02574455)对 529 名不可切除的局部晚期或转移性三阴性乳腺癌 (mTNBC) 患者进行了疗效评估,这些患者在接受过至少两次乳腺癌化疗后复发(一次如果进展发生在 12 个月内,其中可能处于新辅助或辅助环境中)。所有患者均在辅助、新辅助或晚期接受过紫杉烷类药物治疗,除非在第一个紫杉烷类药物周期期间或结束时存在对紫杉烷类药物的禁忌症或不耐受。在招募已知或疑似脑转移患者之前,需要进行磁共振成像 (MRI) 以确定脑转移。脑转移患者被允许在 ASCENT 研究中登记最多 15% 的预先定义的患者。患有已知吉尔伯特病或仅骨病的患者被排除在外。
患者被随机分配 (1:1) 在 21 天 (n=267) 或医生选择的单药化疗 (n=262) 的第 1 天和第 8 天接受 TRODELVY 10 mg/kg 静脉输注。单药化疗由研究者在随机化之前从以下选择之一中确定:艾日布林 (n=139)、卡培他滨 (n=33)、吉西他滨 (n=38) 或长春瑞滨 (n=52)。
患者接受治疗直至疾病进展或出现不可接受的毒性。主要疗效结果是基线时无脑转移(即 BMNeg)的患者的无进展生存期(PFS),这是通过使用实体瘤反应评估标准(RECIST)v1.1 标准评估的盲法、独立、集中审查来衡量的。其他疗效指标包括整个人群(所有有和没有脑转移的患者)的 PFS 和总生存期 (OS)。
整个人群 (n = 529) 患者的中位年龄为 54 岁(范围:27 至 82 岁);99.6%为女性;79% 是白人,12% 是黑人/非裔美国人;81% 的患者年龄 < 65 岁。所有患者的 ECOG 体能状态均为 0 (43%) 或 1 (57%)。42% 的患者有肝转移,9% 为 BRCA1/BRCA2 突变状态阳性,70% 在诊断时为 TNBC。12% 的基线脑转移先前接受过治疗并且稳定(n=61;TRODELVY组 32 例,单药化疗组 29 例)。总体而言,29% 的患者之前接受过 PD-1/PD-L1 治疗。在整个人群中,TRODELVY组中 13% 的患者在转移性环境中仅接受过 1 种先前的全身治疗。
疗效结果总结在表 11 中,并显示在图 1 和图 2 中。在转移性环境中仅接受过 1 线全身治疗的患者亚组的疗效结果(除了在 12 年内出现疾病复发或进展之外)个月的新辅助/辅助全身治疗)与那些在转移环境中至少接受过两种先前治疗的患者一致。
表 11:ASCENT 的疗效结果
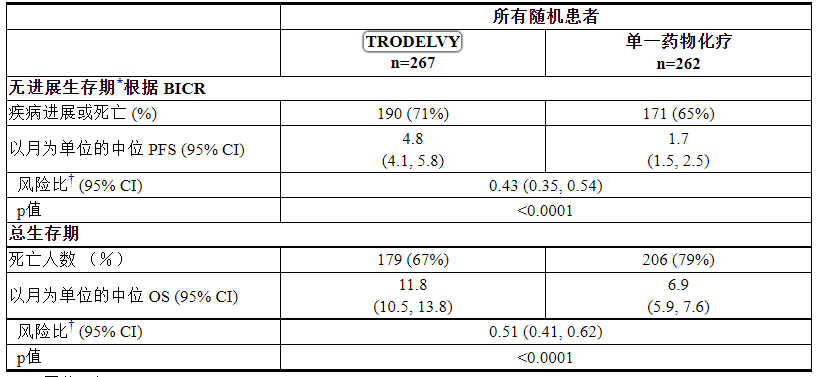
CI = 置信区间
*PFS 定义为从随机化日期到第一次放射学疾病进展或因任何原因死亡的日期,以先到者为准。
†针对分层因素调整的分层对数秩检验:先前化疗的次数、研究开始时已知脑转移的存在和区域。
图 1:在 ASCENT 中通过 BICR(所有随机患者)得出的 PFS 的 Kaplan-Meier 图
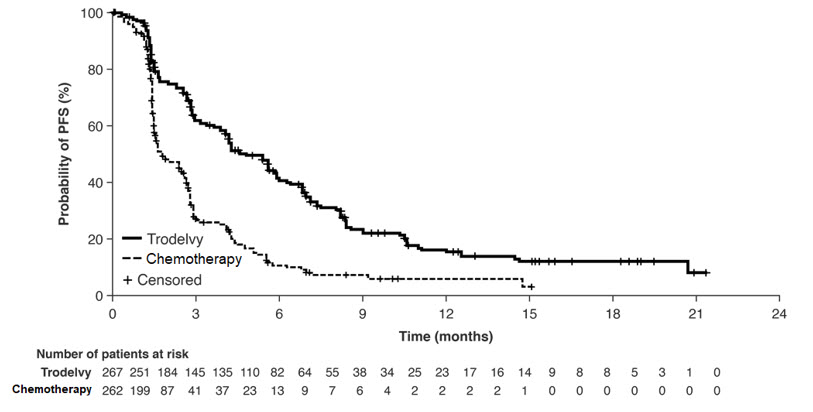
图 2:ASCENT 中 OS(所有随机患者)的 Kaplan-Meier 图
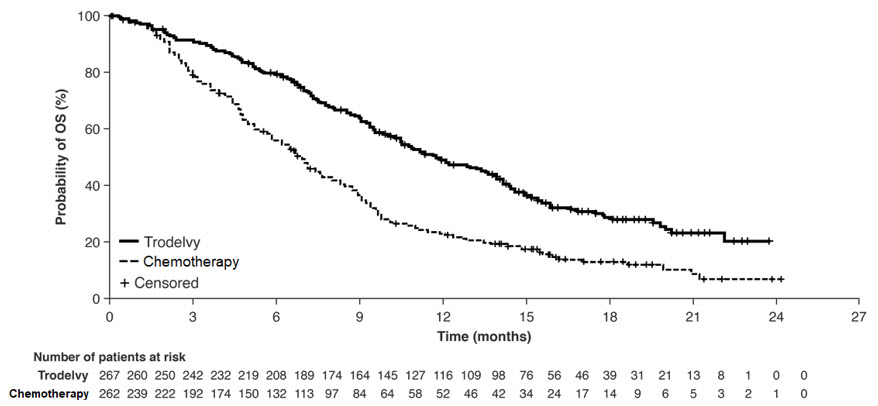
对先前接受过治疗的稳定脑转移患者的 PFS 探索性分析显示分层 HR 为 0.65(95% CI:0.35,1.22)。TRODELVY组的中位 PFS为 2.8 个月(95% CI:1.5、3.9),单药化疗的中位 PFS 为 1.6 个月(95% CI:1.3、2.9)。同一人群的探索性 OS 分析显示分层 HR 为 0.87(95% CI:0.47,1.63)。TRODELVY组的中位 OS为 6.8 个月(95% CI:4.7,14.1),单药化疗的中位 OS 为 7.4 个月(95% CI:4.7,11.1)。
IMMU-132-01
TRODELVY的疗效在一项多中心、单组研究 (NCT01631552) 中得到评估,该研究招募了 108 名转移性三阴性乳腺癌 (mTNBC) 患者,这些患者之前至少接受过两种针对转移性疾病的抗癌治疗。患有大块疾病(定义为大于 7 厘米的肿块)的患者不符合条件。至少 4 周未接受高剂量类固醇(> 20 mg 泼尼松或等效药物)治疗的脑转移患者符合条件。患有已知吉尔伯特病的患者被排除在外。
患者在 21 天治疗周期的第 1 天和第 8 天静脉内接受TRODELVY 10 mg/kg。患者接受TRODELVY治疗,直至疾病进展或对治疗不耐受。每 8 周进行一次肿瘤成像,并在初始部分或完全缓解后 4-6 周进行确认性 CT/MRI 扫描,直至进展需要停止治疗。主要疗效结果指标是研究者使用 RECIST 1.1 评估的总体反应率 (ORR) 和反应持续时间。
中位年龄为 55 岁(范围:31 至 80 岁);87% 的患者年龄小于 65 岁。大多数患者是女性 (99%) 和白人 (76%)。在进入研究时,所有患者的 ECOG 体能状态均为 0 (29%) 或 1 (71%)。76% 有内脏疾病,42% 有肝转移,56% 有肺/胸膜转移,2% 有脑转移。12 名患者 (11%) 在初次诊断时患有 IV 期疾病。
在转移情况下接受过的既往全身治疗的中位数为 3(范围:2 至 10)。转移环境中的既往化疗包括卡铂或顺铂 (69%)、吉西他滨 (55%)、紫杉醇或多西紫杉醇 (53%)、卡培他滨 (51%)、艾日布林 (45%)、多柔比星 (24%)、长春瑞滨 (16) %)、环磷酰胺 (19%) 和伊沙匹隆 (8%)。
总体而言,98% 的患者既往接受过紫杉烷类药物治疗,86% 的患者既往接受过蒽环类药物治疗(新)辅助治疗或转移治疗。
表 12 总结了功效结果。
表 12:IMMU-132-01 中 mTNBC 患者的疗效结果
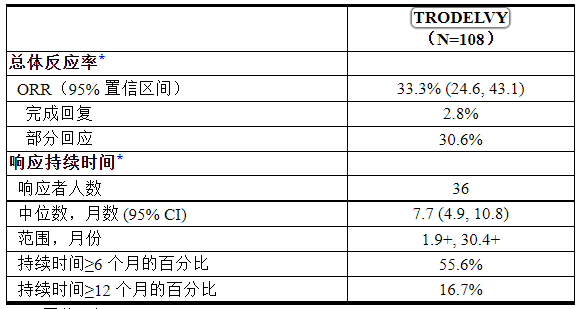
CI:置信区间
+:表示正在进行
*研究者评估
14.2 局部晚期或转移性 HR-阳性、HER2-阴性乳腺癌
TROPiCS-02 研究
TRODELVY的疗效在一项多中心、开放标签、随机研究(TROPiCS-02;NCT03901339)中进行了评估,该研究在 543 名无法切除的局部晚期或转移性 HR 阳性、HER2 阴性(IHC 0、IHC 1+ 或 IHC 2+)患者中进行/ISH–) 乳腺癌,其疾病在以下任何情况下发生进展:CDK 4/6 抑制剂、内分泌治疗和紫杉烷;患者在转移性环境中至少接受过两种既往化疗(如果在 12 个月内复发,其中一种可能是新辅助或辅助化疗)。
患者被随机分配 (1:1),在 21 天周期 (n=272) 的第 1 天和第 8 天接受TRODELVY 10 mg/kg 静脉输注或单药化疗 (n=271)。单药化疗由研究者在随机化之前从以下选择之一中确定:艾日布林 (n=130)、长春瑞滨 (n=63)、吉西他滨 (n=56) 或卡培他滨 (n=22)。根据以下因素对随机化进行分层:转移性疾病的既往化疗方案(2 对 3-4)、内脏转移(是或否)以及在转移情况下至少 6 个月的内分泌治疗(是或否)。
患者接受治疗直至疾病进展或出现不可接受的毒性。如果患者临床稳定并被研究者认为获得临床益处,则允许TRODELVY的给药超出 RECIST 定义的疾病进展。主要疗效结果指标是根据 RECIST v1.1 由 BICR 确定的 PFS。其他疗效指标包括 OS、BICR 的 ORR 和 BICR 的 DOR。
研究人群中患者的中位年龄为 56 岁(范围:27-86 岁),26% 的患者年龄在 65 岁或以上。大多数患者为女性(99%);67% 是白人,4% 是黑人,3% 是亚裔,26% 是未知种族。患者在任何情况下接受过 7 次(范围:3 至 17 次)既往全身化疗方案,在转移情况下接受过 3 次(范围:0 至 8 次)既往全身化疗方案。大约 42% 的患者有 2 种既往化疗方案用于治疗转移性疾病,而 58% 的患者有 3 至 4 种既往化疗方案,并且所有患者的 ECOG 体能状态为 0 (45%) 或 1 (55%)。百分之九十五的患者有内脏转移。大多数患者在转移性环境中接受内分泌治疗 ≥ 6 个月 (86%)。
与单药化疗相比, TRODELVY在 PFS 和 OS 方面表现出统计学上的显着改善。
功效结果总结于表13和图3和图4中。
表 13:TROPiCS-02 的功效结果
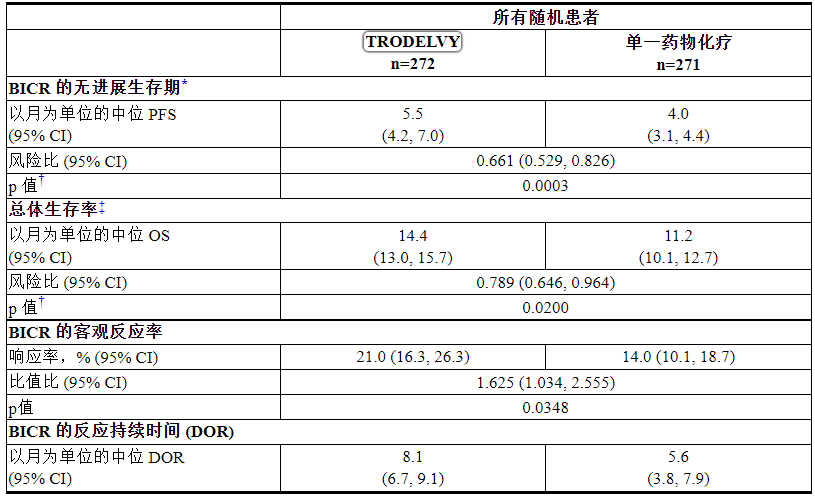
*PFS 定义为从随机化日期到第一次放射学疾病进展或因任何原因死亡的日期,以先到者为准。
†针对分层因素调整的分层对数秩检验:转移性疾病的既往化疗方案(2 对 3-4)、内脏转移(是/否)和转移环境中的内分泌治疗至少 6 个月(是或否) .
BICR = 盲法独立中央审查;CI = 置信区间
‡第二次中期 OS 分析(在观察到 390 次 OS 事件时进行)
图 3:TROPiCS-02 中 BICR 的 PFS 卡普兰-迈尔图
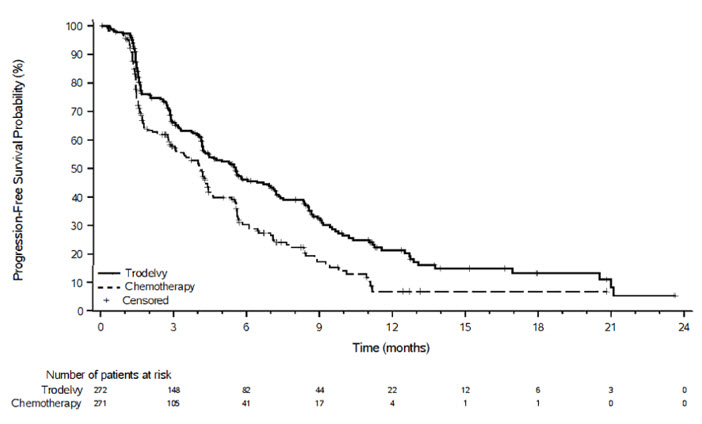
图 4:TROPiCS-02 中 OS 的 Kaplan-Meier 图
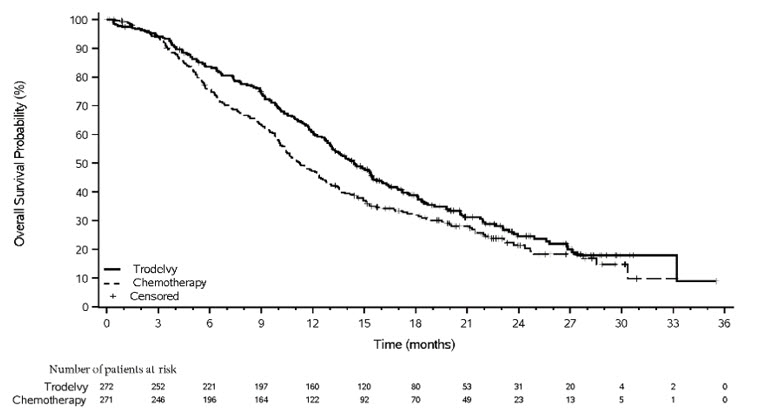
14.3 局部晚期或转移性尿路上皮癌
奖杯研究
TRODELVY的疗效在 TROPHY(NCT03547973)中进行了评估,这是一项单臂、多中心研究,招募了 112 名局部晚期或 mUC 患者,这些患者之前接受过含铂化疗和 PD-1 或 PD-L1 抑制剂治疗。在 21 天治疗周期的第 1 天和第 8 天,患者接受TRODELVY 10 mg/kg 静脉输注。在给予TRODELVY之前,所有患者都接受了预防化疗引起的恶心、呕吐和输液反应的治疗。患者接受治疗直至疾病进展或出现不可接受的毒性。
中位年龄为 66 岁(范围:33 至 90 岁),78% 为男性,74% 为白人,3% 为亚裔,3% 为黑人,20% 未知。所有患者的基线东部肿瘤合作组 (ECOG) 体能状态为 0 (28%) 或 1 (72%)。百分之九十六的患者有转移性疾病;67%的患者有内脏转移,其中34%有肝转移。
既往全身治疗的中位数为 3(范围:1 至 8)。65% 的患者既往接受过顺铂,21% 的患者既往接受过卡铂,13% 的患者既往接受过顺铂和卡铂,100% 的患者既往接受过 PD-1 或 PD-L1 抑制剂。对于 34% 的患者,含铂化疗仅在新辅助/辅助环境中接受。9% 的患者之前接受过 enfortumab vedotin。
主要疗效结果指标是 ORR 和 DOR,由独立审查评估使用实体瘤反应评估标准 (RECIST) v1.1 标准进行评估。表 14 总结了功效结果。
表 14:TROPHY 中局部晚期或 mUC 患者的疗效结果
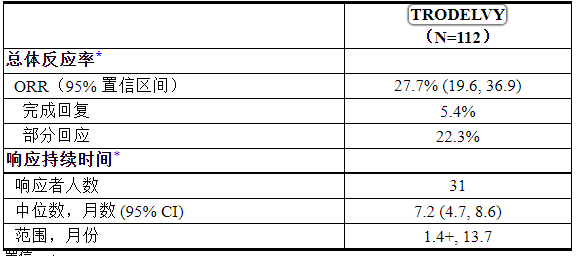
置信区间
+表示正在进行
*通过独立审查评估
15 参考文献
1.“OSHA 危险药物”。职业安全与健康。http://www.osha.gov/SLTC/hazardousdrugs/index.html。
16 如何供应/储存和处理
注射用TRODELVY (sacituzumab govitecan-hziy) 是一种无菌、灰白色至淡黄色冻干粉末,装在单剂量小瓶中。每个TRODELVY小瓶都单独装在纸箱中:
- NDC 55135-132-01 包含一个 180 mg 小瓶
将小瓶储存在 2°C 至 8°C(36°F 至 46°F)的冰箱中,原装纸箱中避光,直至重新配制。不要冻结。
TRODELVY是一种危险药物。遵循适用的特殊处理和处置程序1。
17 患者咨询信息
建议患者阅读 FDA 批准的患者标签(患者信息)
中性粒细胞减少症
忠告患者中性粒细胞减少症的风险。指导患者如他们经受发烧,发冷,或其他感染体征立即联系他们的卫生保健提供者[见警告和注意事项(5.1) ]。
腹泻
忠告患者腹泻的风险。指导患者如果在治疗期间首次出现腹泻,请立即联系他们的医疗保健提供者;黑色或血便;脱水症状,如头晕、头晕或昏厥;由于恶心或呕吐而无法口服液体;或无法在 24 小时内控制腹泻[见警告和注意事项 (5.2) ]。
超敏反应和输液相关反应
告知患者严重输注反应和过敏反应的风险。指导患者如果在治疗期间出现面部、嘴唇、舌头或喉咙肿胀、荨麻疹、呼吸困难、头晕、头晕、发冷、寒战、喘息、瘙痒、潮红、皮疹、低血压或发烧,立即联系他们的医疗保健提供者输注后 24 小时内[见警告和注意事项 (5.3) ]。
恶心,呕吐
忠告患者恶心和呕吐的风险。还建议根据既定指南使用两种或三种药物方案进行术前用药,以预防化疗引起的恶心和呕吐 (CINV)。还可以根据临床指示使用额外的止吐药、镇静剂和其他支持措施。所有患者都应获得带回家的药物,以预防和治疗迟发性恶心和呕吐,并附有明确的说明。指导患者如他们经受不受控制的恶心或呕吐立即联系他们的卫生保健提供者[见警告和注意事项(5.4) ]。
胚胎-胎儿毒性
忠告女性患者如她们怀孕或成为怀孕联系她们的卫生保健提供者。告知女性患者对胎儿的风险和潜在流产[见特殊人群中使用(8.1) ]。
避孕
建议有生殖潜力的女性患者在治疗期间和末次服用TRODELVY后 6 个月内使用有效的避孕措施 [见在特定人群中的使用(8.3) ]。
建议有生殖潜能女性伴侣的男性患者在治疗期间和末次服用TRODELVY 后 3 个月内使用有效的避孕措施[见在特定人群中的使用(8.3) ]。
哺乳期
建议女性在治疗期间和末次服用TRODELVY 后 1 个月内不要母乳喂养[见在特定人群中使用 (8.2) ]。
不育症
建议具有生殖潜力的女性TRODELVY可能损害生育能力[见在特定人群中的使用(8.3) ]。
制造商:
Gilead Sciences, Inc.
333 Lakeside Dr.
Foster City, CA 94404, USA
美国许可证号 2258

主要显示面板 - 180 mg 瓶标签
NDC 55135-132-01
仅限处方药
TRODELVY ®
sacituzumab govitecan-hziy
注射用
每瓶 180 毫克
仅用于静脉输液
警告:危险药物
单剂量小瓶
丢弃未使用的部分
90370103

主要展示面板 - 180 毫克小瓶盒
NDC 55135-132-01
仅限处方药
TRODELVY ®
sacituzumab govitecan-hziy
注射用
每瓶 180 毫克
仅用于静脉输液
警告:危险药物
使用前立即重新配制和稀释
单剂量小瓶
丢弃未使用的部分
1 瓶

【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
本说明书来源于:美国FDA网站
https://nctr-crs.fda.gov/fdalabel/services/spl/set-ids/57a597d2-03f0-472e-b148-016d7169169d/spl-doc?hl=TRODELVY
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书。



PRINCIPAL DISPLAY PANEL - 180 mg Vial Label
NDC 55135-132-01
Rx only
TRODELVY®
sacituzumab govitecan-hziy
For injection
180 mg per vial
For intravenous infusion only
Warning: Hazardous Drug
Single-dose vial
Discard unused portion
90370103

PRINCIPAL DISPLAY PANEL - 180 mg Vial Box
NDC 55135-132-01
Rx only
TRODELVY®
sacituzumab govitecan-hziy
For injection
180 mg per vial
For intravenous infusion only
Warning: Hazardous Drug
Reconstitute and dilute
immediately prior to use
Single-dose vial
Discard unused portion
1 Vial
【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
-

通用名: 注射用戈沙妥珠单抗
商品名: TRODELVY
规格: 180mg
产地: BSP制药股份有限公司(BSP Pharmaceuticals S.p.A.) 新加坡
国际参考零售价:¥**/瓶
-