商品详情
返回产品目录
商品包装及说明书因厂家更换频繁,如有不符以实物为主
注射用恩美曲妥珠单抗
国际零售参考价:¥**/瓶
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。
完整的处方信息
警告:肝毒性,心脏毒性,胚胎安全性毒性
肝毒性:已报道严重的肝毒性,包括接受KADCYLA治疗的患者的肝衰竭和死亡。在开始KADCYLA治疗之前和每次KADCYLA剂量之前,监测血清转氨酶和胆红素。如果血清转氨酶升高或总胆红素升高,则酌情降低剂量或停用KADCYLA。(2.3,5.1)
心脏毒性: 施用KADCYLA可能导致左心室射血分数(LVEF)降低。在KADCYLA治疗之前和期间评价所有患者的左心室功能。对于左心室功能的临床显着下降,暂不接受治疗。(2.3,5.2)
胚胎-胎儿毒性:怀孕期间暴露于KADCYLA会导致胚胎-胎儿伤害。告知患者这些风险以及有效避孕的必要性。(5.3,8.1,8.3)
1适应症和用途
1.1转移性乳腺癌(MBC)
KADCYLA ®,作为单一药剂,被指示为谁先前接收到的曲妥珠单抗和紫杉烷,单独地或结合HER-2阳性转移性乳腺癌的患者的治疗。患者应具有:
◎曾经接受过转移性疾病的治疗,或
◎完成辅助治疗期间或六个月内出现疾病复发。
根据FDA批准的KADCYLA伴随诊断选择患者进行治疗[ 参见剂量和用法(2.1) ]。
1.2早期乳腺癌(EBC)
KADCYLA作为一种单一药物,可用于辅助治疗HER2阳性早期乳腺癌的患者,这些患者在新辅助紫杉烷和曲妥珠单抗治疗后仍残留浸润性疾病。
根据FDA批准的KADCYLA伴随诊断选择患者进行治疗[ 参见剂量和用法(2.1) ]。
2用法用量
2.1患者选择
根据肿瘤样本中的HER2蛋白过表达或HER2基因扩增选择患者[参见适应症和用法(1),临床研究(14) ]。 HER2蛋白过表达和/或HER2基因扩增的评估应使用经FDA认可的专门针对乳腺癌的检测方法,并应由具有熟练水平的实验室进行。有关FDA批准的用于检测HER2蛋白过表达和HER2基因扩增的检测的信息,请访问:http://www.fda.gov/CompanionDiagnostics。
化验性能不当,包括使用次优固定组织,未能使用指定的试剂,偏离特定化验说明以及未能包括用于化验验证的适当对照,均可能导致结果不可靠。
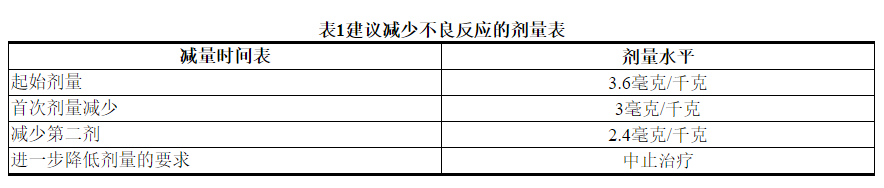
2.2建议剂量和时间表
请勿将曲妥珠单抗替代或与KADCYLA替代。
每3周(21天周期)静脉输注KADCYLA的推荐剂量为3.6 mg / kg。不要以大于3.6 mg / kg的剂量施用KADCYLA。
在给药过程中密切监测输液部位是否有皮下浸润[见警告和注意事项(5.9) ]。
第一次输注:在90分钟内进行输注。在输注过程中以及初始剂量后至少90分钟内观察患者的发烧,发冷或其他与输注相关的反应[请参阅警告和注意事项(5.5) ]。
后续输注:如果先前的输注耐受性良好,则应进行30分钟以上的给药。在输液期间以及输液后至少30分钟观察患者。
转移性乳腺癌(MBC)
MBC患者应接受治疗,直到疾病进展或无法控制的毒性。
早期乳腺癌(EBC)
除非疾病复发或无法控制的毒性,否则EBC患者应接受总共14个周期的治疗。
2.3剂量修改
减少剂量后,请勿重新增加KADCYLA剂量。
如果延迟或错过了计划的剂量,请尽快给药;不要等到下一个计划的周期。调整给药计划,以维持两次给药之间的3周间隔。以患者在最近一次输液中耐受的剂量和速率进行输液。
如果患者出现与输注相关的反应,请减慢或中断KADCYLA的输注速度。永久终止KADCYLA以进行危及生命的输液相关反应[请参阅警告和注意事项(5.5) ]。
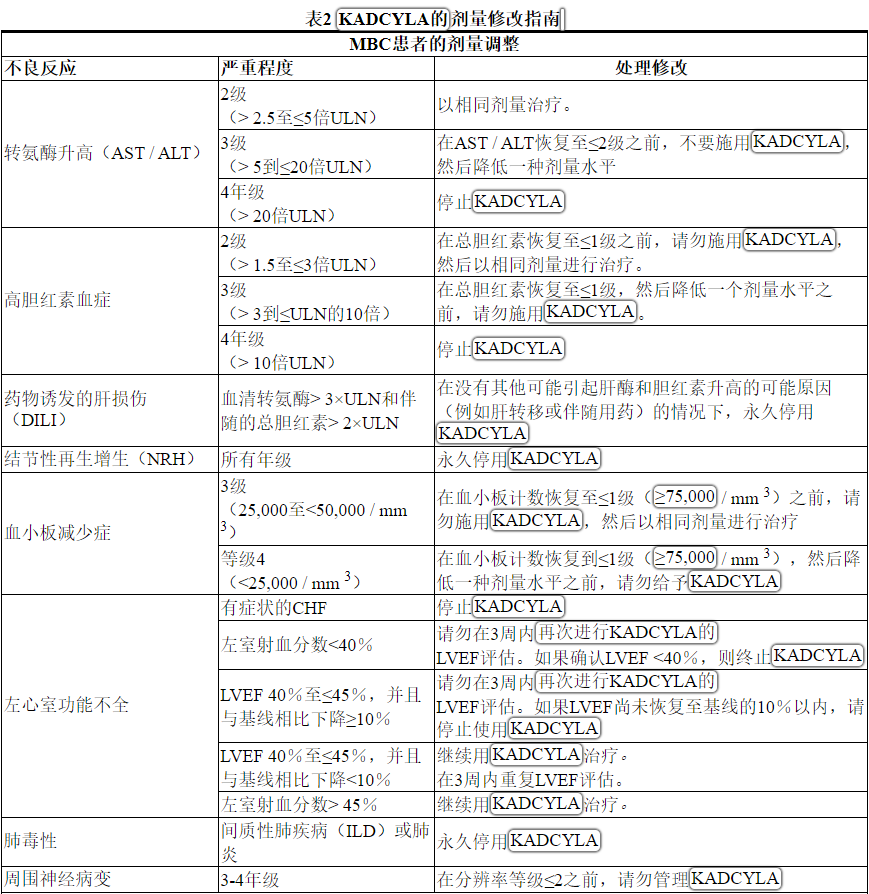
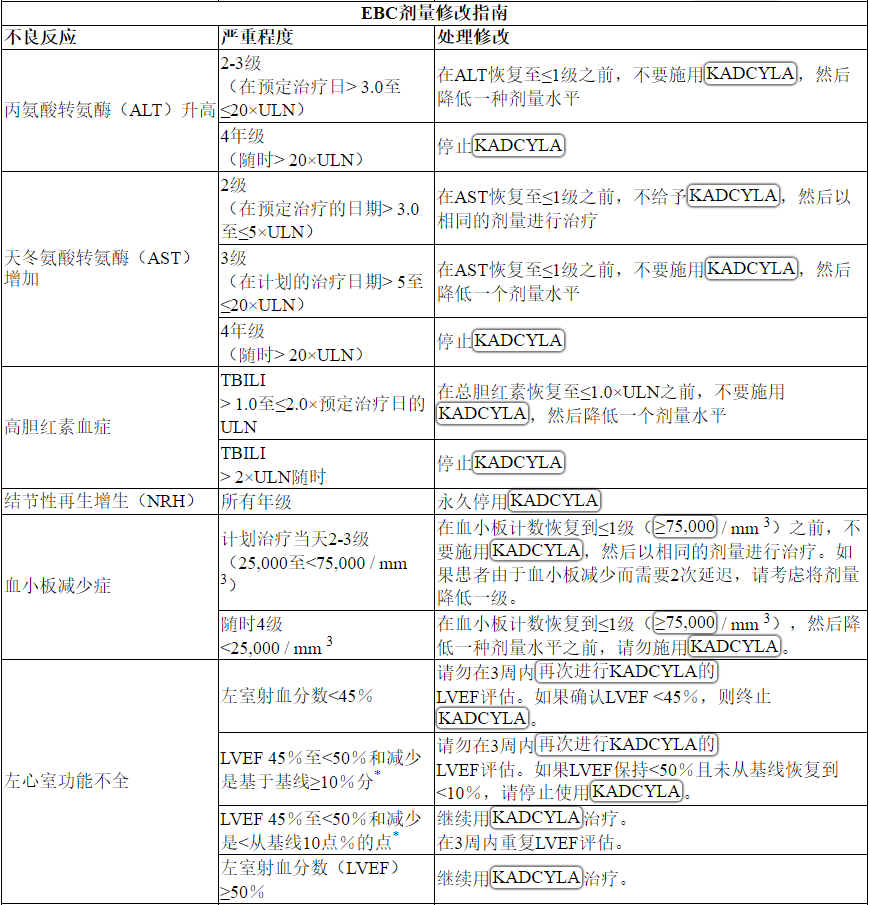
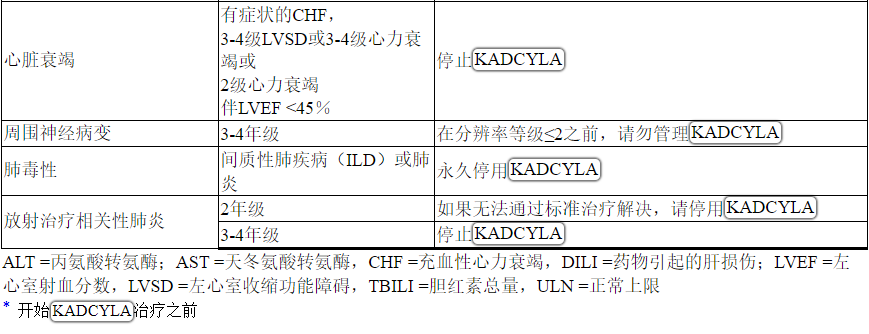
根据表1和表2提供的指南,应对血清转氨酶升高,高胆红素血症,左心室功能障碍,血小板减少,肺毒性或周围神经病变的治疗可能需要暂时中断,降低剂量或中止KADCYLA。
2.4管理准备
为了防止用药错误,重要的是要检查小瓶标签,以确保正在准备和给药的药物是KADCYLA(阿托曲妥珠单抗坦坦碱)而不是曲妥珠单抗。
管理:
◎辖KADCYLA静脉输液只用0.2或0.22微米的线聚醚砜(PES)过滤器。请勿静脉推注或推注。
◎请勿将KADCYLA与其他药物混合使用或作为输液剂使用。
重构:
◎使用无菌技术来重构和制备定量溶液。应使用适当的化学治疗药物制备程序。
◎使用无菌注射器,向100 mg KADCYLA小瓶中缓慢注入5 mL注射用无菌水,或向160 mg KADCYLA小瓶中缓慢注入8 mL注射用无菌水,以生成含20 mg / mL的溶液。轻轻旋转小瓶,直到完全溶解。 不要摇晃。检查重新配制的溶液中是否有颗粒和变色。
◎重新配制的溶液应澄清至微乳白色且无可见颗粒。重构溶液的颜色应为无色至浅棕色。如果重新配制的溶液含有可见的颗粒或浑浊或变色,请勿使用。
◎配制的冻干小瓶在用无菌注射用水配制后应立即使用。如果不立即使用,重构的KADCYLA小瓶可以在2ºC至8ºC(36°F至46°F)的冰箱中保存24小时。24小时后丢弃未使用的KADCYLA。 不要冻结。
◎重构产品不含防腐剂,仅用于单剂量。
稀释:
确定KADCYLA 的正确剂量(mg)[请参阅剂量和用法(2.1) ]。
◎计算所需的20 mg / mL重组KADCYLA溶液的体积。
◎从小瓶中取出此量,并将其添加到装有250 mL 0.9%氯化钠注射液的输液袋中。 不要使用葡萄糖(5%)溶液。
◎轻轻地将袋子倒置以混合溶液,以避免起泡。
◎稀释的KADCYLA输注溶液应立即使用。如果不立即使用,可在使用前将溶液在2°C至8°C(36°F至46°F)的冰箱中保存24小时。该存储时间不包括重构小瓶允许的时间。 请勿冻结或摇动。
3剂型和强度
单剂量小瓶中的冻干粉:每小瓶100 mg或每小瓶ado-曲妥珠单抗emtansine 160 mg。
4禁忌症
没有。
5警告和注意事项
5.1肝毒性
在使用KADCYLA的 临床试验中已观察到肝毒性,主要表现为无症状,瞬时升高的血清转氨酶浓度[见不良反应(6.1) ]。以KADCYLA为单药的临床试验(n = 1624)中已观察到严重的肝毒性,包括3例致命死亡。所有致命病例均发生在KADCYLA的 MBC临床试验中,其中包括严重的药物诱发的肝损伤和相关的肝性脑病。一些经历肝毒性的患者有合并症和/或伴随药物,具有已知的肝毒性潜能。
在开始KADCYLA治疗之前和每次KADCYLA剂量之前,监测血清转氨酶和胆红素。患有已知活动性肝病(例如,乙型肝炎病毒或丙型肝炎病毒)的患者被排除在EMILIA和KATHERINE研究之外[见临床研究(14.1) ]。如果血清转氨酶升高和/或总胆红素升高,则酌情减少剂量或中止KADCYLA [见剂量和给药方法(2.2) ]。对于血清转氨酶> 3×ULN并伴有总胆红素> 2×ULN的患者,永久停用KADCYLA治疗。 卡迪拉 在开始治疗之前,尚未对血清转氨酶> 2.5×ULN或胆红素> 1.5×ULN的患者进行研究。
在KADCYLA的临床试验中,已从肝活检中鉴定出肝结节性再生增生(NRH)病例(1624例接受治疗的患者中有5例死亡,其中之一是致命的)。这5例NRH病例中有2例在EMILIA中观察到,而2例在KATHERINE中观察到[请参阅不良反应(6.1) ]。NRH是一种罕见的肝脏疾病,其特征是肝实质广泛地良性转化为小的再生结节。NRH可能导致非肝硬化门脉高压。NRH的诊断只能通过组织病理学来确认。对于所有在肝电脑断层扫描(CT)扫描中发现有门静脉高压和/或类似肝硬化样征但临床转氨酶正常且无其他肝硬化表现的患者,应考虑NRH。诊断为NRH后,必须永久终止KADCYLA治疗。
5.2左心室功能不全
用KADCYLA治疗的患者发生左心室功能障碍的风险增加。在KADCYLA治疗的患者中,LVEF降至<40%。在KADCYLA的临床试验中已观察到严重的心力衰竭病例,没有致命病例。在EMILIA,KADCYLA治疗组中有1.8%的患者发生左心功能不全,拉帕替尼加卡培他滨治疗组中有3.3%的患者发生左心功能不全。在KATHERINE中,在KADCYLA治疗组中有0.4%的患者发生左心功能不全,在曲妥珠单抗治疗组中有0.6%的患者发生左心功能不全[见不良反应(6.1) ]。
在开始KADCYLA之前和治疗期间应定期(例如,每三个月)评估LVEF,以确保LVEF在机构的正常范围内。在开始治疗前,LVEF <50%的患者尚未研究过用KADCYLA治疗。
对于MBC患者,在常规监测下,如果LVEF <40%或40%至45%,且绝对值低于治疗前值的10%或更多,则保留KADCYLA,并在约3周内重复进行LVEF评估。如果LVEF没有改善或进一步下降,则永久停用KADCYLA。
对于EBC患者,在常规监测下,如果LVEF <45%或为45%至49%,且绝对值低于治疗前值的10%或更多,则不接受KADCYLA,并在大约3周内重复进行LVEF评估。如果LVEF尚未改善或进一步下降,则永久停用KADCYLA [请参阅剂量和用法(2.2) ]。
有症状充血性心力衰竭(CHF),严重心律不齐或有6个月以内心肌梗塞或不稳定型心绞痛病史的患者被排除在EMILIA和KATHERINE研究之外[见临床研究(14.1) ]。
5.3胚胎-胎儿毒性
向孕妇服用时,KADCYLA可能会造成胎儿伤害。在上市后的环境中,接受曲妥珠单抗(KADCDYLA的抗体成分)治疗的患者观察到羊水过少和羊水过少的序列表现为肺发育不全,骨骼异常和新生儿死亡。DM1是KADCYLA的细胞毒性成分,基于其作用机理,可引起胚胎-胎儿毒性。
在开始KADCYLA之前,验证具有生殖潜能的女性的怀孕状况。建议孕妇和有生殖能力的女性在怀孕期间或在怀孕前七个月内接触KADCYLA可能会造成胎儿伤害。繁殖潜力的指教女性治疗期间和7个月内使用有效避孕的最后剂量后KADCYLA [见特殊人群中使用(8.1,8.3) ]。
5.4肺毒性
在KADCYLA的临床试验中,已报告了包括肺炎在内的间质性肺病(ILD)病例,其中一些导致急性呼吸窘迫综合征或致命的结局。体征和症状包括呼吸困难,咳嗽,疲劳和肺部浸润。
在MBC患者中,据报道肺炎的发生率为0.8%(884名接受治疗的患者中有7名),其中1例为3级肺炎。在EMILIA,肺炎的总发病率为1.2%。据报道,在KATHERINE中,肺炎的发生率为1.1%(740例接受KADCYLA治疗的患者中有8例),其中1例为3级肺炎。
据报道,放射性肺炎的发生率为1.8%(623例接受辅助放疗和KADCYLA治疗的患者中有11例),其中2例为3级放射性肺炎[见不良反应(6.1) ]。
对于确诊为ILD或肺炎的患者,永久停止使用KADCYLA治疗。对于辅助环境下的放射性肺炎患者,对于≥3级或对标准治疗无反应的2级,应永久停用KADCYLA [请参阅剂量修改(2.2) ]。
由于晚期恶性肿瘤,合并症和同时进行肺部放射治疗而导致的休息时呼吸困难的患者发生肺毒性的风险可能增加。
5.5输注相关反应,超敏反应
对于因输注相关反应(IRR)和/或超敏反应而永久停用曲妥珠单抗的患者,尚未研究过KADCYLA的治疗。治疗KADCYLA不建议对这些患者。
在KADCYLA的临床试验中,已报道了与输注相关的反应,其特征在于以下症状中的一种或多种:潮红,畏寒,发热,呼吸困难,低血压,喘息,支气管痉挛和心动过速。在EMILIA,接受KADCYLA治疗的患者中IRR的总发生率为1.4%。在KATHERINE中,接受KADCYLA治疗的患者IRR的总发生率为1.6%[见不良反应(6.1) ]。在大多数患者中,输注结束后的数小时至一天中,这些反应消失了。 重度IRR患者应中断KADCYLA治疗。 卡迪拉如果发生危及生命的IRR,则应永久终止治疗[见剂量和用法(2.2) ]。应密切观察患者的IRR反应,尤其是在第一次输注期间。
在单药KADCYLA的临床试验中已观察到一例严重的过敏/过敏样反应。治疗此类反应的药物以及应急设备应可立即使用。
5.6出血
在KADCYLA的临床试验中,已经报道了包括中枢神经系统,呼吸道和胃肠道出血在内的出血事件。这些出血事件中的一些导致致命的后果。在EMILIA,KADCYLA治疗组出血总发生率为32%,拉帕替尼加卡培他滨治疗组为16%。≥3级出血的发生率在KADCYLA治疗组中为1.8%,在拉帕替尼联合卡培他滨治疗组中为0.8%。在KATHERINE中,KADCYLA治疗组出血总发生率为29%,曲妥珠单抗治疗组出血总发生率为10%。KADCYLA≥3级出血的发生率为0.4%治疗组,其中1例死亡为颅内出血,曲妥珠单抗治疗组为0.3%[见不良反应(6.1) ]。尽管在某些观察到的病例中,患者也正在接受抗凝治疗,抗血小板治疗或患有血小板减少症,但在其他情况下,则没有已知的其他危险因素。请谨慎使用这些药物,并在医学上需要同时使用时考虑进行其他监测。
5.7血小板减少
在KADCYLA的临床试验中报告了血小板减少症或血小板计数减少(16 24例≥3级的患者中有145例;1624≥3级的患者中有494例)。这些患者中的大多数发生1级或2级事件(
在EMILIA,KADCYLA治疗组的血小板减少症总发生率为31%,拉帕替尼加卡培他滨治疗组的血小板减少率为3.3%[见不良反应(6.1) ]。≥3级血小板减少症的发生率在KADCYLA治疗组为15%,拉帕替尼加卡培他滨治疗组为0.4%。在亚洲患者中,KADCYLA治疗组的≥3级血小板减少症的发生率为45%,拉帕替尼加卡培他滨治疗的组为1.3%。
在KATHERINE中,血小板减少症的总发生率在KADCYLA治疗组为29%,在曲妥珠单抗治疗组为2.4%[见不良反应(6.1) ]。≥3级血小板减少症的发生率在KADCYLA治疗组中为6%,在曲妥珠单抗治疗组中为0.3%。在亚洲患者中,KADCYLA治疗组中≥3级血小板减少症的发生率为19%,曲妥珠单抗治疗组中为0%。KADCYLA治疗组中亚洲患者血小板减少症的总发生率为50%。
在开始KADCYLA之前和每次KADCYLA剂量之前监测血小板计数[请参阅剂量和给药方法(2.2) ]。 在开始治疗前,血小板计数<100,000 / mm 3的患者尚未研究KADCYLA。在事件的血小板减少到级≥3(<50,000 /毫米3)不管理KADCYLA直到血小板计数恢复到1级(≥75000个/ mm 3)[见剂量和给药方法(2.2) ]。密切监测血小板减少症(<100,000 / mm 3)和使用KADCYLA治疗期间接受抗凝治疗的患者。
5.8神经毒性
在KADCYLA的临床试验中报告了周围神经病变,主要表现为1级,主要是感觉觉(1624例≥3级的患者中有26例;任何级别的1624例患者中有435例)。在EMILIA,KADCYLA治疗组的周围神经病变总发生率为21%,拉帕替尼加卡培他滨治疗组为14%[见不良反应(6.1) ]。在KADCYLA治疗组中,≥3级周围神经病变的发生率为2.2%,在拉帕替尼联合卡培他滨治疗组中为0.2%。在KATHERINE,周围神经病变的总发病率是32%在KADCYLA治疗组和曲妥珠单抗治疗组中的17%。对于接受KADCYLA治疗的患者,周围神经病变(包括感觉和运动周围神经病变)在进行KATHERINE的主要IDFS分析时,尚无30%的病例得到解决。≥3级周围神经病变的发生率在KADCYLA治疗组为1.6%,曲妥珠单抗治疗组为0.1%。
在出现3级或4级周围神经病的患者中,应暂时停用KADCYLA,直到病情降至≤2级。应持续监测患者的神经毒性体征或症状[参见非临床毒理学(13.2) ]。
5.9外渗
在KADCYLA临床研究中,观察到了继发于外渗的反应。这些反应在输注后24小时内更频繁地观察到,通常是轻度的,包括在输注部位出现红斑,压痛,皮肤刺激,疼痛或肿胀。KADCYLA外渗的具体治疗方法尚不清楚。给药期间应密切监测输液部位是否有皮下浸润。
6不良反应
标签的其他部分详细讨论了以下不良反应:
◎肝毒性[请参阅警告和注意事项(5.1) ]
◎左心室功能障碍[请参阅警告和注意事项(5.2) ]
◎胚胎胎儿毒性[请参阅警告和注意事项(5.3) ]
◎肺毒性[请参阅警告和注意事项(5.4) ]
◎输注相关反应,超敏反应[请参阅警告和注意事项(5.5) ]
◎出血[请参阅警告和注意事项(5.6) ]
◎血小板减少症[请参阅警告和注意事项(5.7) ]
◎神经毒性[请参阅警告和注意事项(5.8) ]
6.1临床试验经验
由于临床试验是在广泛不同的条件下进行的,因此无法将在某种药物的临床试验中观察到的不良反应率直接与另一种药物的临床试验中观察到的不良反应率进行比较,并且可能无法反映实际中观察到的不良反应率。
警告和注意事项中的数据反映了1624名患者每3周(21天周期)静脉输注KADCYLA单一药物3.6 mg / kg的情况,其中包括884例HER2阳性转移性乳腺癌患者和740例患者HER2阳性早期乳腺癌(KATHERINE试验)。
转移性乳腺癌
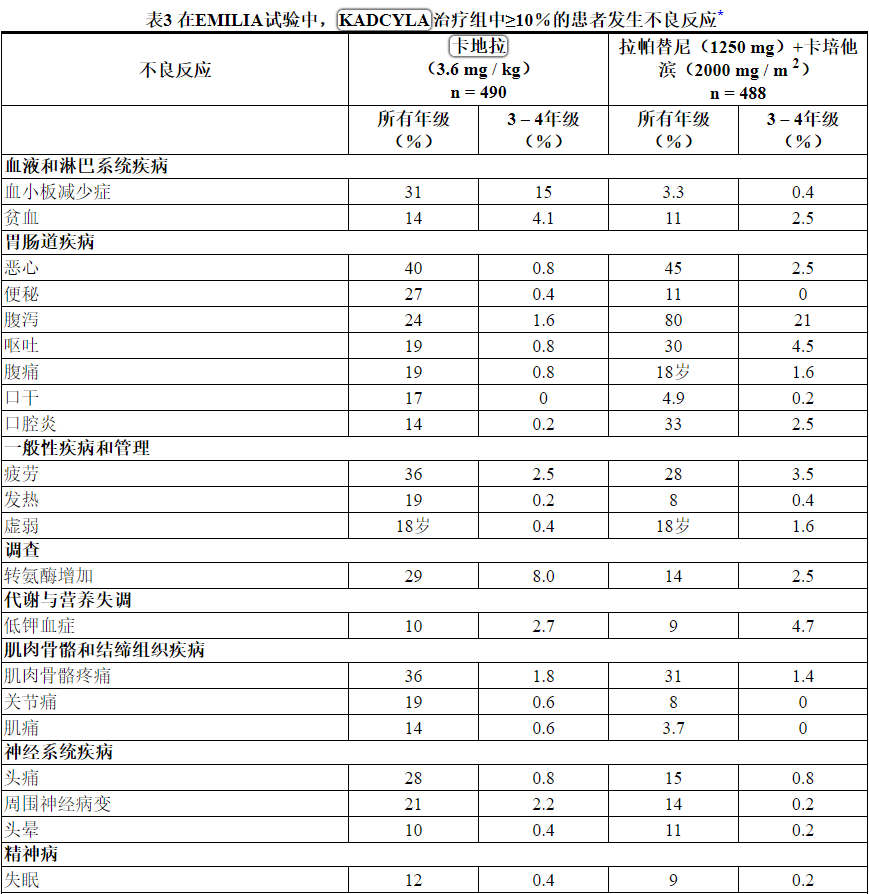
在临床试验中,KADCYLA已被评估为884例HER2阳性转移性乳腺癌患者的单药治疗。最常见的不良反应(≥25%)为疲劳,恶心,肌肉骨骼疼痛,出血,血小板减少症,头痛,转氨酶升高,便秘和鼻st。
表3中描述的不良反应已在EMILIA试验中治疗的HER2阳性转移性乳腺癌患者中确定[见临床研究(14.1) ]。患者被随机分配接受KADCYLA或拉帕替尼加卡培他滨治疗。KADCYLA治疗组患者的研究治疗中位时间为7.6个月,拉帕替尼和卡培他滨治疗的患者分别为5.5个月和5.3个月。
在EMILIA试验中,KADCYLA治疗组中有43%的患者出现≥3级不良反应,而拉帕替尼加卡培他滨治疗组中则有59%的患者发生不良反应。
允许调整KADCYLA的剂量[参见剂量和用法(2.2) ]。32例患者(7%)因不良反应而停用KADCYLA,而41例患者(8%)因不良反应而停用拉帕替尼和51例患者(10%)因不良反应而停用卡培他滨。导致KADCYLA停用的最常见不良反应是血小板减少症和转氨酶升高。接受KADCYLA治疗的80位患者(16%)出现不良反应,导致剂量降低。导致KADCYLA剂量减少的最常见不良反应(≥1%的患者)包括血小板减少症,转氨酶升高和周围神经病。导致剂量延迟的不良反应发生在116位(24%)KADCYLA治疗的患者中。导致KADCYLA剂量延迟的最常见不良反应(≥1%的患者)是中性粒细胞减少症,血小板减少症,白细胞减少症,疲劳,转氨酶升高和发热。
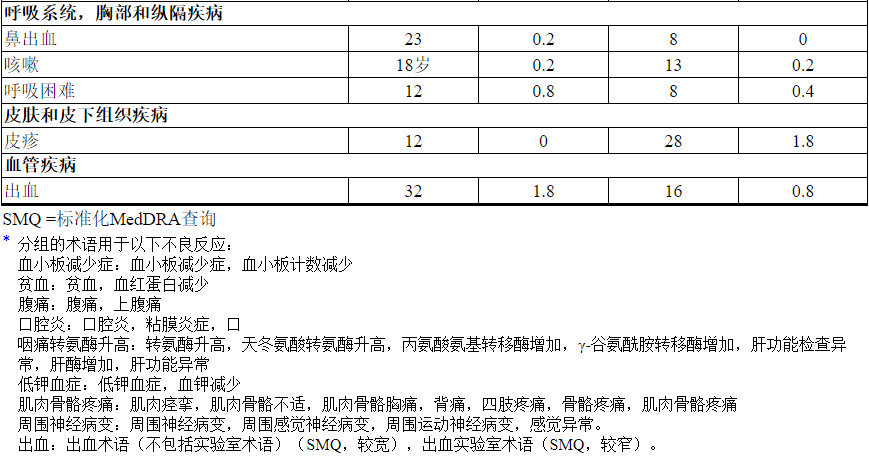
表3报告了在EMILIA试验的KADCYLA治疗组(n = 490)的患者中发生的不良反应。所选实验室异常见表4。在随机试验中,KADCYLA最常见的不良反应(频率> 25%)为恶心,疲劳,肌肉骨骼疼痛,出血,血小板减少,转氨酶升高,头痛和便秘。最常见的NCI–CTCAE(第3版)≥3级不良反应(频率> 2%)是血小板减少症,转氨酶升高,贫血,低钾血症,周围神经病变和疲劳。
在EMILIA ,经KADCYLA治疗组的患者中,以下临床不良反应报告为少于10%:消化不良(9%),尿路感染(9%),畏寒(8%),消化不良(8%),中性粒细胞减少(7%),周围水肿(7%),瘙痒(6%),高血压(5%),血碱性磷酸酶升高(4.7%),视力模糊(4.5%),结膜炎(3.9%),干眼症(3.9 %),流泪增加(3.3%),药物超敏反应(2.2%),左心功能不全(1.8%),输液相关反应(1.4%),肺炎(1.2%),结节性再生增生(0.4%),门脉高压(0.4%)。
早期乳腺癌
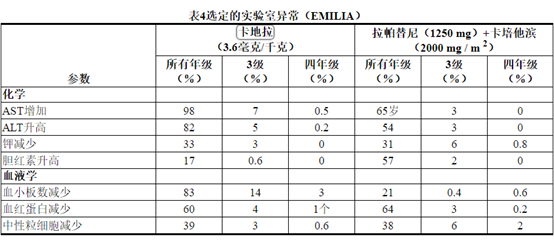
KADCYLA已被评估为740例HER2阳性早期乳腺癌患者的单药治疗。
在表5中描述的不良反应已在KATHERINE试验中治疗的HER2阳性早期乳腺癌患者中确定[见临床研究(14.2) ]。患者被随机分配接受KADCYLA或曲妥珠单抗治疗。KADCYLA治疗组的患者的研究治疗中位时间为10个月,曲妥珠单抗治疗的患者为10个月。
KADCYLA治疗组中有190名(26%)患者经历了≥3级不良反应,而曲妥珠单抗组中有111名(15%)患者。133名患者(18%)因不良反应而停用KADCYLA,而15名患者(2.1%)因不良反应而停用曲妥珠单抗。
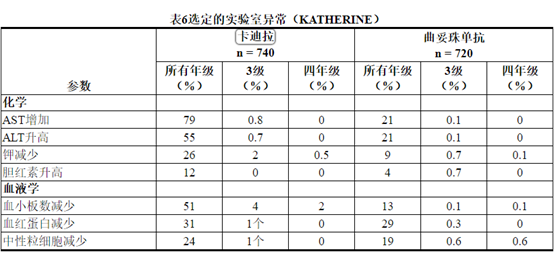
导致KADCYLA停用的最常见不良反应(≥1%的患者)是血小板计数降低,血胆红素升高,射血分数降低,AST升高,ALT升高和周围神经病。
允许调整KADCYLA的剂量[参见剂量和用法(2.2) ]。用KADCYLA治疗的一百零六名患者(14%)剂量减少。导致KADCYLA剂量降低的最常见不良反应(≥1%的患者)包括血小板减少症,转氨酶升高,血液胆红素和疲劳。106名(14%)接受KADCYLA治疗的患者发生了导致剂量延迟的不良反应。导致KADCYLA剂量延迟的最常见不良反应(≥1%的患者)是中性粒细胞减少,血小板减少和AST增加。
所选实验室异常见表6。在随机试验中,KADCYLA最常见的不良反应(频率> 25%)为疲劳,恶心,转氨酶升高,肌肉骨骼疼痛,出血,血小板减少症,头痛,周围神经病变和关节痛。NCI–CTCAE(第3版)最常见的≥3级不良反应(> 2%)是血小板减少症和高血压。
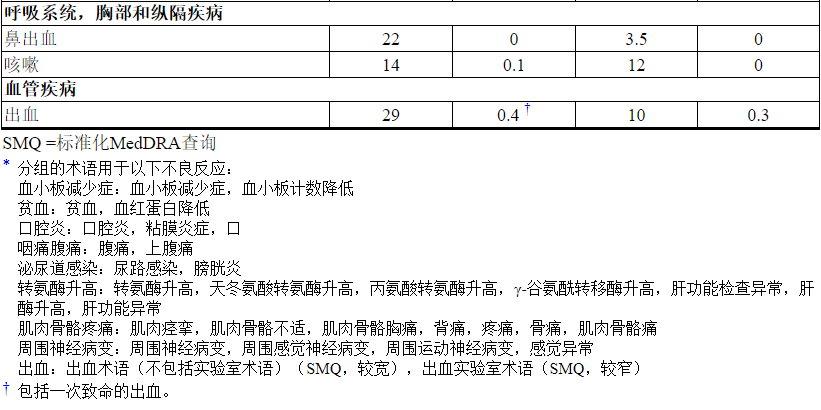
在KATHERINE 的KADCYLA治疗组中,报告了<10%的患者以下临床相关不良反应:血液碱性磷酸酶升高(8%),消化不良(8%),呼吸困难(8%),中性粒细胞减少症(8%),血液胆红素增加(7%),低血钾(7%),瘙痒(7%),高血压(6%),流泪增加(6%),畏寒(5%),干眼症(4.5%),消化不良(4.3%) ),周围水肿(3.9%),视力模糊(3.9%),结膜炎(3.5%),左心功能不全(3.0%),药物超敏反应(2.7%),输液相关反应(1.6%),放射性肺炎(1.5) %),肺炎(1.1%),皮疹(1.1%),乏力(0.4%),结节性再生增生(0.3%)。
6.2免疫原性
与所有治疗性蛋白质一样,对KADCYLA可能产生免疫反应。在多个时间点对来自七个临床研究的1243名患者进行了针对KADCYLA的抗药物抗体(ADA)反应的测试。以下KADCYLA的患者给药,5.1%(1243分之63),用于抗测试为阳性KADCYLA在一个或多个给药后的时间点的抗体。在临床研究中,有6.4%(24/376)的患者抗KADCYLA抗体检测为阳性。在EMILIA,有5.2%(24/466)的患者抗KADCYLA抗体检测为阳性,其中13例中和抗体也呈阳性。在KATHERINE中,3.7%(15/401)的患者抗KADCYLA测试呈阳性抗体,其中5个也对中和抗体呈阳性。由于ADA的发生率低,因此无法就抗KADCYLA抗体对KADCYLA的药代动力学,安全性和功效的影响得出结论。ADA采样时患者血清中KADCYLA的存在可能会干扰该检测抗KADCYLA抗体的能力。结果,数据可能无法准确反映抗KADCYLA抗体发育的真实发生率。
免疫原性数据高度依赖于所用测试方法的敏感性和特异性。此外,在测试方法中观察到的阳性结果发生率可能会受到多种因素的影响,包括样品处理,样品采集时间,药物干扰,伴随用药和潜在疾病。因此,将KADCYLA抗体的发生率与其他产品的抗体发生率进行比较可能会产生误导。抗KADCYLA抗体的临床意义尚不清楚。
6.3上市后经验
在批准后使用KADCYLA的过程中,已经确认了以下不良反应。由于这些反应是从不确定大小的人群中自愿报告的,因此并非总是能够可靠地估计其发生频率或建立与药物暴露的因果关系。
◎肿瘤溶解综合征(TLS):用KADCYLA治疗的患者已报告可能的TLS病例。具有明显肿瘤负担(例如,大转移)的患者可能处于较高的风险中。患者可能出现高尿酸血症,高磷酸盐血症和急性肾功能衰竭,这可能代表可能的TLS。提供者应考虑临床指示的其他监测和/或治疗。
7药物相互作用
尚未与KADCYLA进行正式的药物相互作用研究。 体外研究表明,KADCYLA的细胞毒性成分DM1 主要通过CYP3A4代谢,而在较小程度上被CYP3A5代谢。应避免将强效CYP3A4抑制剂(例如,酮康唑,伊曲康唑,克拉霉素,阿扎那韦,茚地那韦,奈法唑酮,奈非那韦,利托那韦,沙奎那韦,替利霉素和伏立康唑)与KADCYLA并用1,因为这可能增加毒性并增加其毒性。考虑使用另一种没有或抑制CYP3A4的药物。如果不可避免地需要同时使用强效CYP3A4抑制剂,请考虑延迟KADCYLA直至可能的情况下,将强效CYP3A4抑制剂从循环中清除(约3种抑制剂的半衰期)。如果同时使用强效CYP3A4抑制剂且不能延迟KADCYLA治疗,则应密切监测患者的不良反应。
8在特定人群中的使用
8.1怀孕
怀孕药物警戒计划
有一个针对KADCYLA的怀孕药物警戒计划。如果在怀孕期间服用KADCYLA,或者患者在接受KADCYLA时或在最后一次服用KADCYLA后7个月内怀孕,则医疗服务提供者和患者应立即致电 1-888-835-2555向Genentech 报告暴露于KADCYLA。
风险摘要
向孕妇服用时,KADCYLA可能会造成胎儿伤害。没有关于孕妇使用KADCYLA的可用数据。在上市后的设定中观察到与曲妥珠单抗治疗的抗体组分的患者表现为肺发育不良,骨骼异常,和新生儿死亡的羊水过少和羊水过少序列例KADCYLA [见数据 ]。基于其作用机理,当将KADCYLA的DM1成分施用于孕妇时,也会引起胚胎胎儿的伤害[ 参见数据 ]。告知患者胎儿的潜在风险。如果存在KADCYLA,则有临床考虑在孕妇中使用,或如果患者在最后一次服用KADCYLA 后7个月内怀孕[请参见临床注意事项 ]。
对于指定人群,估计的主要先天缺陷和流产的背景风险尚不清楚。在美国普通人群中,临床公认的怀孕中主要先天缺陷和流产的估计背景风险分别为2-4%和15-20%。
临床注意事项
胎儿/新生儿不良反应
谁收到监测妇女KADCYLA怀孕期间或妊娠羊水过少前7个月内。如果发生羊水过少,请进行适合胎龄并符合社区护理标准的胎儿测试。
数据
人数据
没有关于孕妇使用KADCYLA的可用数据。在上市后的环境中,在妊娠期间使用曲妥珠单抗治疗后,观察到羊水过少和羊水过少的病例,在胎儿中表现为肺发育不全,骨骼异常和新生儿死亡。这些病例报告描述了接受曲妥珠单抗单独或联合化疗的孕妇羊水过少。在某些病例报告中,曲妥珠单抗停用后羊水指数增加。在一种情况下,曲妥珠单抗治疗在羊膜指数改善后恢复,羊水过少复发。
动物资料
没有关于用ado-曲妥珠单抗emtansine进行的生殖和发育毒理学研究。DM1,KADCYLA的细胞毒性成分,破坏微管功能。DM1对快速分裂的动物细胞具有毒性,并且具有遗传毒性,这表明它具有引起胚胎毒性和致畸性的潜力。在器官形成期间,对怀孕的食蟹猕猴给予曲妥珠单抗的研究最高剂量为25 mg / kg,每周两次(临床剂量的7倍),曲妥珠单抗在早期(妊娠第20至50天)越过了胎盘屏障。和妊娠后期(妊娠120至150天)。曲妥珠单抗在胎儿血清和羊水中的浓度分别约为母体血清中浓度的33%和25%,但与不良的发育影响无关。
8.2哺乳
风险摘要
没有关于人乳中存在ado-曲妥珠单抗emtansine,对母乳喂养婴儿的影响或对牛奶产量的影响的信息。DM1是KADCYLA的细胞毒性成分,基于其作用机理,可能会在母乳喂养婴儿中引起严重的不良反应[参见数据 ]。劝告妇女在治疗期间以及最后一次服用KADCYLA后 7个月内不要母乳喂养。
数据
没有用ado-曲妥珠单抗Emtansine或KADCYLA(DM1)的细胞毒性成分进行的动物泌乳研究。在哺乳期食蟹猕猴中,曲妥珠单抗在母乳喂养之前(从妊娠120天开始)和产后(从产后28天开始)以每周两次(每次25 mg / kg的剂量)存在于母乳中,约占母体血清浓度的0.3%。约为KADCYLA临床剂量的7倍)。从出生到1个月大时,血清曲妥珠单抗含量可检测的婴儿猴子对生长或发育没有任何不良影响。
8.3生殖潜力的男性和女性
验孕
在开始KADCYLA之前,验证具有生殖潜能的女性的怀孕状况。
避孕
女性
怀孕期间服用KADCYLA可能对胚胎胎儿造成伤害。劝告有生殖潜力的女性在治疗期间以及最后一次服用KADCYLA后 7个月内使用有效的避孕方法[ 见在特定人群中使用(8.1) ]。
雄性
由于具有潜在的遗传毒性,建议具有生殖潜力的女性伴侣的男性患者在用KADCYLA治疗期间以及最后一次用药后的4个月内使用有效的避孕方法。
不孕症
根据动物毒性研究的结果,KADCYLA可能会损害雌性和雄性生殖能力。目前尚不清楚这种作用是否可逆[见非临床毒理学(13.1) ]。
8.4小儿使用
尚未在儿童患者中确定KADCYLA的安全性和有效性。
8.5老年人使用
在EMILIA中被随机分配至KADCYLA的495例患者中(见临床研究(14.1)),年龄≥65岁的患者65例(13%),年龄≥75岁的患者11例(2%)。≥65岁的患者(两个治疗组的n = 138),无进展生存期(PFS)和总体生存期(OS)的危险比分别为1.06(95%CI:0.68,1.66)和1.05(95%CI: 0.58、1.91)。年龄≥65岁的患者与年龄<65岁的患者相比,KADCYLA的安全性没有总体差异。EMILIA没有包括足够多的75岁以上的患者以得出有关KADCYLA在该年龄组中的安全性或有效性的结论。
在KATHERINE中随机分配至KADCYLA的743例患者中[参见临床研究(14.2) ],年龄≥65岁的患者58例(8%),年龄≥75岁的患者2例(0.3%)。年龄≥65岁的患者与年龄<65岁的患者相比,KADCYLA的安全性或有效性没有总体差异。KATHERINE没有纳入足够的≥75岁患者,无法得出该年龄组中KADCYLA的安全性或有效性的结论。
群体药代动力学分析表明,年龄对ado-曲妥珠单抗emtansine的药代动力学没有临床意义的影响[见临床药理学(12.3) ]。
8.6肾功能不全
尚未进行针对KADCYLA的专门肾功能损害试验。根据人群的药代动力学以及对3级以上不良反应和剂量调整的分析,对于轻度(肌酐清除率[CLcr] 60至89 mL / min)或中度(CLcr 30)患者,无需调整KADCYLA剂量至59 mL / min)肾功能不全。由于可获得的数据有限,建议不建议对严重肾功能不全(CLcr小于30 mL / min)的患者进行剂量调整[见临床药理学(12.3) ]。
8.7肝功能不全
轻度或中度肝功能不全的患者无需调整起始剂量[见临床药理学(12.3) ]。 没有对严重肝功能不全患者进行KADCYLA研究。密切监测因使用KADCYLA观察到的已知肝毒性而导致肝功能不全的患者 [ 见警告和注意事项,肝毒性(5.1) ]。
10过量
目前尚无过量的KADCYLA解毒剂。在临床试验中,据报道过量的KADCYLA约为推荐剂量的两倍,导致2级血小板减少(4天后解决)并导致一名死亡。在致命的情况下,该患者错误地接受了6 mg / kg的KADCYLA,并在服药过量约3周后死亡。尚未确定死亡原因以及与KADCYLA的因果关系。
11说明
KADCYLA(ado-trastuzumab emtansine)是靶向HER2的抗体-药物偶联物(ADC),包含通过稳定的硫醚接头MCC(共价连接至微管抑制药物DM1(美登素衍生物)的人源化抗HER2 IgG1曲妥珠单抗)。 4- [N-马来酰亚胺基甲基]环己烷-1-甲酸)。Emtansine是指MCC-DM1复合物。
曲妥珠单抗是由哺乳动物(中国仓鼠卵巢)细胞产生的特征明确的重组单克隆抗体产品,小分子成分(DM1和MCC)是通过化学合成产生的。Ado-曲妥珠单抗Emtansine每个抗体平均含有3.5个DM1分子。Ado-曲妥珠单抗Emtansine具有以下化学结构:
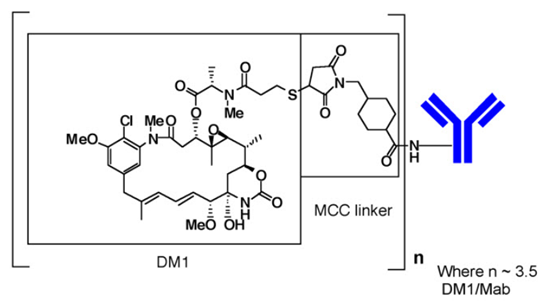
注意:括号中的结构是DM1加MCC,代表了鞣酸成分。每个曲妥珠单抗(Mab)分子平均n为3.5个DM1分子。
KADCYLA(阿托曲妥珠单抗丹参碱)是单剂量小瓶中无菌的白色至灰白色防腐剂游离冻干粉。每个小瓶包含100毫克或160毫克的阿妥曲妥单抗Emtansine。重建后,每个单剂量小瓶均含阿托曲妥珠单抗(20 mg / mL),聚山梨酯20 [0.02%(w / v)],琥珀酸钠(10 mM)和蔗糖[6%(w / v)]。 pH值为5.0。稀释后通过静脉输注施用含有20mg / mL的ado-曲妥珠单抗氨丹宁的所得溶液。
12临床药理学
12.1行动机制
Ado-曲妥珠单抗emtansine是靶向HER2的抗体-药物偶联物。该抗体是人源化抗HER2 IgG1曲妥珠单抗。小分子细胞毒素DM1是微管抑制剂。与HER2受体的亚结构域IV结合后,ado-曲妥珠单抗Emtansine经历受体介导的内在化和随后的溶酶体降解,从而导致含DM1的细胞毒性代谢产物在细胞内释放。DM1与微管蛋白的结合会破坏细胞中的微管网络,从而导致细胞周期停滞和凋亡性细胞死亡。此外,体外 研究表明,与曲妥珠单抗类似,ado-曲妥珠单抗氨丹宁可抑制HER2受体信号转导,介导抗体依赖性细胞介导的细胞毒性,并抑制过度表达HER2的人乳腺癌细胞中HER2胞外域的脱落。
12.2药效学
心脏电生理学
在51名HER2阳性转移性乳腺癌患者的开放标签单臂研究中,评估了多剂量KADCYLA(每3周3.6 mg / kg)对QTc间隔的影响。在研究中未发现平均QT间隔(即> 20 ms)的大变化。
12.3药代动力学
在一项1期研究和一项针对ado-曲妥珠单抗emtansine缀合物(ADC)的人群药代动力学分析中,使用来自5项乳腺癌患者试验的汇总数据评估了KADCYLA的药代动力学。线性两室模型具有从中央室中消除一阶的能力,足以描述ADC浓度-时间曲线。除ADC外,还测定了总抗体(缀合和未缀合的曲妥珠单抗)DM1的药代动力学。ADC的群体药代动力学分析表明,根据疾病状况(佐剂与转移性情况),KADCYLA暴露无差异。KADCYLA的药代动力学总结如下。
分配
接近输注即将结束时,观察到ADC和DM1的最大浓度(C max)。在EMILIA,平均(SD)ADC和DM1周期图1C 最大值以下KADCYLA给药为83.4(16.5)μg/ mL和4.61(1.61)分别纳克/毫升,。在KATHERINE,平均(SD)ADC和DM1周期图1C 最大值以下KADCYLA给药为72.6(24.3)μg/ mL和4.71(2.25)分别纳克/毫升,。
在体外,DM1与人血浆蛋白的平均结合率为93%。在体外, DM1是P-糖蛋白(P-gp)的底物。
根据群体药代动力学分析,ADC的中心分布容积为3.13L。
代谢
体外研究表明,KADCYLA的小分子成分DM1 通过CYP3A4 / 5代谢。DM1 在体外不抑制或诱导主要的CYP450酶。 在人血浆中,以低水平检测到阿杜曲妥珠单抗丹参碱代谢产物MCC-DM1,Lys-MCC-DM1和DM1。
消除
根据人群药代动力学分析,静脉输注KADCYLA后,ADC的清除率为0.68 L /天,消除半衰期(t 1/2)约为4天。每3周重复给药一次静脉输注后,未观察到KADCYLA的积聚。
根据群体药代动力学分析(n = 671),体重,RECIST靶病变最长直径的总和,HER2细胞外域(ECD)浓度,AST,白蛋白和基线曲妥珠单抗浓度被确定为ado-trastuzumab的统计学显着协变量精氨酸清除率。但是,这些协变量对ado-曲妥珠单抗氨丹宁暴露的影响程度表明,除体重外,这些协变量不太可能对KADCYLA暴露具有临床意义的影响。因此,以每3周3.6 mg / kg的体重为基础的剂量(不校正其他协变量)被认为是合适的。
肾功能不全的影响
根据对668例患者的人群药代动力学分析,包括中度(CLcr 30-59 mL / min,n = 53)和轻度(CLcr 60-89 mL / min,n = 254)肾功能不全,表明ADC的药代动力学不是与正常肾功能(CLcr≥90 mL / min,n = 361)相比,受轻度至中度肾功能不全的影响。仅提供了一名严重肾功能不全(CLcr <30 mL / min)的患者的数据[见特定人群的使用(8.7) ]。
肝功能不全的影响
肝脏是消除含有DM1和DM1的分解代谢产物的主要器官。在对肝功能正常(n = 10),轻度(Child-Pugh A; n = 10)的转移性HER2阳性乳腺癌患者施用3.6 mg / kg的KADCYLA后,评估了ado-trastuzumab emtansine和含DM1的分解代谢物的药代动力学。 = 10)和中度(Child-Pugh B; n = 8)肝功能不全。
- 含和不含DM1和DM1的分解代谢产物(Lys-MCC-DM1和MCC-DM1)的血浆浓度较低,在有和没有肝功能不全的患者之间相当。
- 轻度和中度肝功能不全患者在第1周期中ado-trastuzumab氨丹宁的全身暴露(AUC)分别比正常肝功能患者低38%和67%。轻度或中度肝功能不全患者在反复给药后第3周期的Ado-曲妥珠单抗氨丹宁(AUC)处于正常肝功能患者的观察范围内。
尚未对患有严重肝功能不全(Child-Pugh C级)的患者进行KADCYLA研究。
年龄和种族的影响
根据人群药代动力学分析,年龄(<65 [n = 577]; 65-75(n = 78);> 75 [n = 16])和种族(亚洲[n = 73];非亚洲[n = 598] ])对ado-trastuzumab氨丹宁的药代动力学没有临床意义的影响。
13毒理学
13.1致癌,诱变,生育力受损
尚未使用ado-曲妥珠单抗emtansine进行致癌性研究。
在体内单剂量大鼠骨髓微核试验中,DM1在造血或裂伤中的暴露量可与给予KADCYLA的人测得的DM1的平均最大浓度相当。DM1 在体外细菌反向突变(Ames)分析中不致突变。
根据动物毒性研究的结果,KADCYLA可能损害人类的生育能力。在ado-trastuzumab emtansine对大鼠的单剂量毒性研究中,在严重毒性剂量水平(60 mg / kg;约为临床暴露量的4倍)下,睾丸生精管变性并伴有睾丸和附睾重量增加。基于AUC)。在雌性大鼠中,相同剂量会导致卵巢出血和黄体坏死的迹象。每三周一次以12周(四剂)的剂量服用ado-trastuzumab氨丹宁的猴子(最高剂量为30 mg / kg(基于AUC的临床暴露量的7倍)),附睾,前列腺,睾丸,精囊和子宫,尽管由于所招募动物的性成熟程度不同,对这些作用的解释尚不清楚。
13.2动物毒理学和/或药理学
在猴子中,使用剂量达30 mg / kg的ado-trastuzumab氨丹宁(基于AUC的临床暴露量的7倍左右)进行治疗,会导致坐骨神经中的剂量依赖性轴突变性,并伴有施旺细胞的肥大或增生,以及轴突变性。脊髓的背侧fun骨。根据细胞毒性成分DM1的作用机理,存在神经毒性的临床潜力[请参阅警告和注意事项(5.8) ]。
14临床研究
14.1转移性乳腺癌
在一项随机,多中心,开放标签试验(EMILIA)(NCT00829166)中对KADCYLA的疗效进行了评估,该试验对991例HER2阳性,不可切除的局部晚期或转移性乳腺癌患者进行了评估。在试验入组前,需要事先使用紫杉烷和曲妥珠单抗为基础的治疗方法。仅接受过辅助治疗的患者必须在完成辅助治疗期间或完成治疗后的六个月内复发疾病。要求乳腺肿瘤样品显示HER2过表达,定义为在中央实验室测定的3+ IHC或FISH扩增比率≥2.0。患者被随机分配(1:1)接受拉帕替尼加卡培他滨或KADCYLA。根据研究者的确定,按世界地区(美国,西欧等),不可切除的局部晚期或转移性疾病的先前化疗方案的数量(0-1,> 1)和内脏与非内脏疾病进行了分层。
在21天周期的第1天,以3.6 mg / kg的剂量静脉内给予KADCYLA。在21天周期的第1至14天中,每天以1250 mg /天口服一次拉帕替尼,并在1000 mg / m 2口服中以每天两次口服卡培他滨。患者接受KADCYLA或拉帕替尼加卡培他滨治疗,直至疾病进展,同意撤消或出现不可接受的毒性。在初级分析的时候,对研究药物的中值时间是5.7个月(范围:0-28.4)为KADCYLA为卡培他滨,4.9个月(范围:0-30.8)拉帕替尼,和4.8个月(范围0-30.4) 。
这项研究的共同主要疗效结果是基于独立审查委员会(IRC)对肿瘤反应的评估的无进展生存期(PFS)和总体生存期(OS)。PFS定义为从随机分组日期到疾病进展或任何原因导致死亡(以较早发生者为准)的时间。总生存期定义为从随机分组日期到任何原因死亡日期之间的时间。其他结果包括PFS(基于研究者的肿瘤反应评估),客观反应率(ORR),反应持续时间和症状发展时间。
治疗组之间患者人口统计学和基线肿瘤特征是平衡的。所有患者在研究开始时均患有转移性疾病。中位年龄约为53岁(范围为24-84岁),白人占74%,亚裔占18%,黑人占5%。除5名患者外,其余均为女性。美国有27%的患者入选,欧洲有32%,亚洲有16%。肿瘤的预后特征包括激素受体状态(阳性:55%,阴性:43%),仅存在内脏疾病(68%)和非内脏疾病(33%)以及转移部位的数量(<3:61%, ≥3:37%)在研究组中相似。
大多数患者(88%)在转移环境中接受过先前的全身治疗。12%的患者仅在新辅助或辅助条件下接受过治疗,并且在治疗后6个月内疾病复发。除一名患者外,所有患者在进入研究前均接受曲妥珠单抗治疗。约有85%的患者在转移性环境中接受过曲妥珠单抗治疗。超过99%的患者接受紫杉烷治疗,而61%的患者在进入研究之前接受了蒽环类抗生素治疗。总体而言,患者在转移环境中接受了3种全身性药物的治疗。在激素受体阳性肿瘤患者中,有44.4%接受过先前的辅助激素治疗,而44.8%的患者接受了局部晚期/转移性疾病的激素治疗。
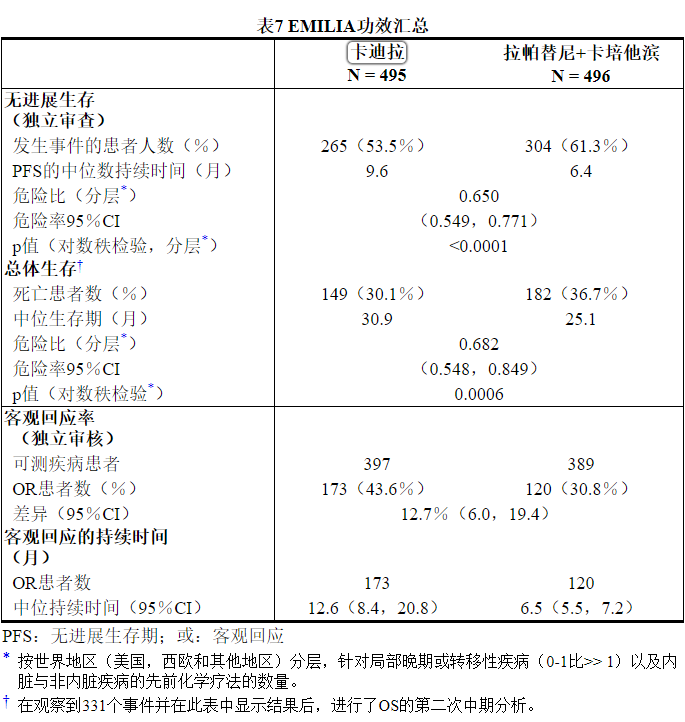
这项随机试验显示,与拉帕替尼联合卡培他滨治疗组相比,KADCYLA治疗组的IRC评估的PFS有统计学上的显着改善[危险比(HR)= 0.65,95%CI:0.55,0.77,p <0.0001], PFS的中位数增加了3.2个月(KADCYLA治疗组的中位PFS为9.6个月,拉帕替尼加卡培他滨组的中位PFS为6.4个月)。见表7和图1。研究者评估的PFS的结果与IRC评估的PFS的结果相似。
在进行PFS分析时,有223例患者死亡。与KADCYLA组(19 %)相比,拉帕替尼联合卡培他滨组发生的死亡更多(26%),但是该中期OS分析的结果未达到预先设定的统计意义上的终止边界。在第二次临时操作系统分析时,发生了331个事件。达到了OS的主要共同终点;接受KADCYLA的患者的OS显着改善(HR = 0.68,95%CI:0.55,0.85,p = 0.0006)。该结果越过了预先设定的功效停止边界(HR = 0.73或p = 0.0037)。KADCYLA组的中位生存期为30.9个月,而拉帕替尼加卡培他滨组的中位生存期为25.1个月。请参阅表7和图2。
KADCYLA的治疗益处根据分层因素,主要基线人口统计学和疾病特征以及以前的治疗,在患者亚组中观察到了PFS和OS方面的差异。在激素受体阴性疾病的患者亚组(n = 426)中,PFS和OS的危险比分别为0.56(95%CI:0.44、0.72)和0.75(95%CI:0.54、1.03)。在激素受体阳性疾病的患者亚组(n = 545)中,PFS和OS的危险比分别为0.72(95%CI:0.58、0.91)和0.62(95%CI:0.46、0.85)。根据IRC评估,在不可测量疾病的亚组(n = 205)中,PFS和OS的危险比分别为0.91(95%CI:0.59,1.42)和0.96(95%CI:0.54,1.68) , 分别; 在可测量疾病患者中,危险比分别为0.62(95%CI:0.52、0.75)和0.65(95%CI:0.51、0.82),分别。65岁以下(n = 853)的患者的PFS和OS危险比分别为0.62(95%CI:0.52,0.74)和0.66(95%CI:0.52,0.83)。≥65岁的患者(n = 138),PFS和OS的危险比分别为1.06(95%CI:0.68,1.66)和1.05(95%CI:0.58,1.91)。
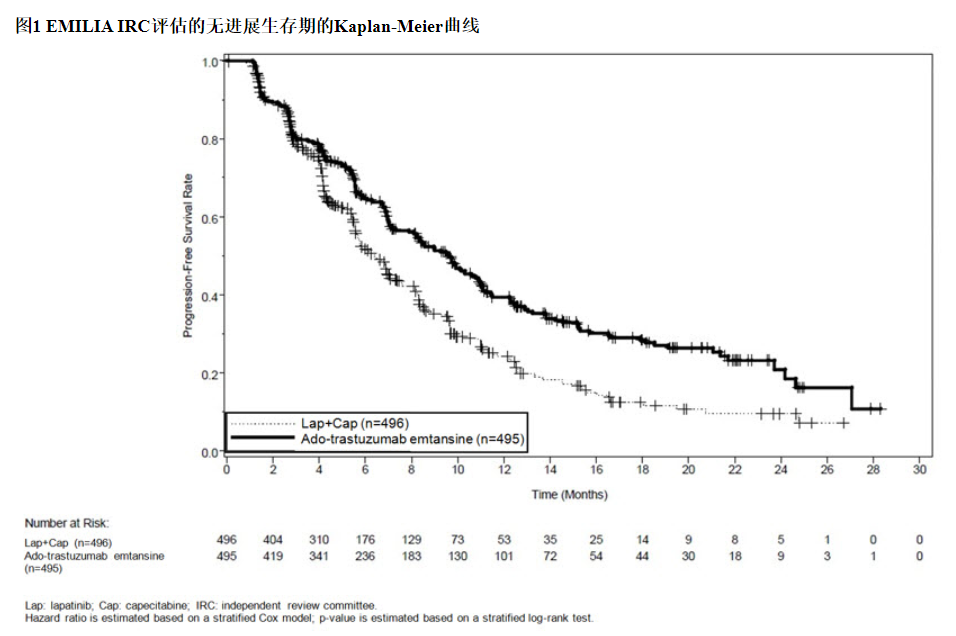
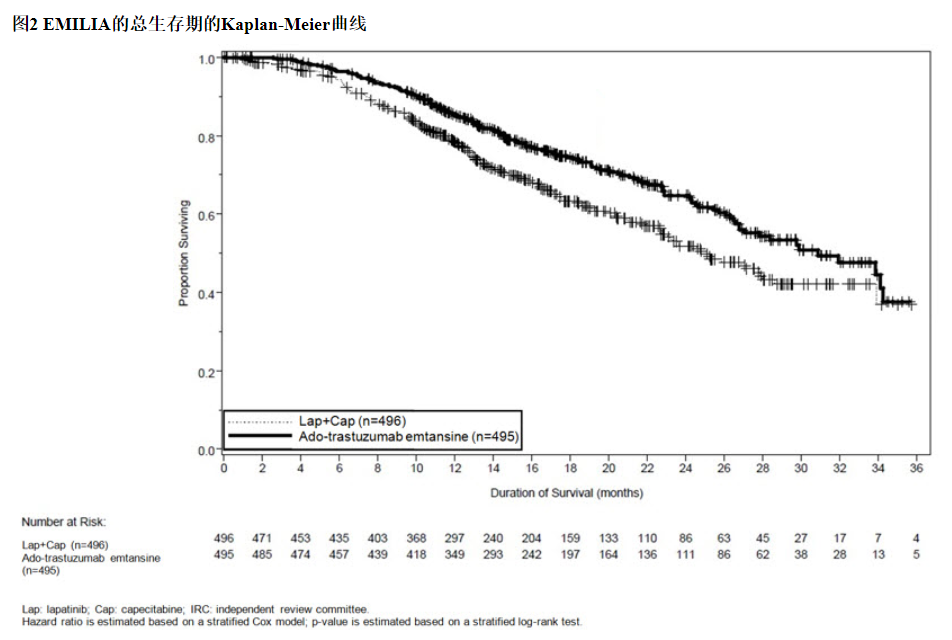
14.2早期乳腺癌
KATHERINE(NCT01772472)是一项针对1486例HER2阳性早期乳腺癌患者的随机,多中心,开放标签试验。患者需要接受新辅助紫杉烷和曲妥珠单抗治疗,并在乳腺和/或腋窝淋巴结中残留浸润性肿瘤。根据当地指南,患者在接受研究治疗的同时接受放射治疗和/或激素治疗。要求乳腺癌样品显示HER2过表达,定义为在3中心实验室使用Ventana的PATHWAY抗HER2- / neu(4B5)兔单克隆一抗或INFORM HER2双ISH DNA探针鸡尾酒检测法测定的HER2过表达定义为3+ IHC或ISH扩增比≥2.0 。患者被随机(1:1)接受KADCYLA或曲妥珠单抗。根据就诊时的临床阶段,激素受体状态,术前HER2指导治疗(曲妥珠单抗,曲妥珠单抗加其他HER2指导药物)和术前治疗后病理性淋巴结状态评估,对患者进行分层。
在21天周期的第1天,以3.6 mg / kg的剂量静脉内给予KADCYLA。在21天周期的第1天,以6 mg / kg的剂量静脉给予曲妥珠单抗。除非出现疾病复发,同意撤回或不可接受的毒性反应,否则接受KADCYLA或曲妥珠单抗治疗的患者共进行了14个周期的治疗。在进行主要疗效结果分析时,KADCYLA-和曲妥珠单抗治疗的患者的中位治疗持续时间均为10个月。出于疾病考虑和研究者的酌情考虑,如果合适,因疾病复发以外的原因而终止KADCYLA的患者可以完成剩余计划的曲妥珠单抗HER2定向治疗方案的其余部分。
该研究的主要功效结果是无创生存期(IDFS)。IDFS定义为从随机分组日期到同侧浸润性乳腺癌复发,同侧局部或区域浸润性乳腺癌复发,远处复发,对侧浸润性乳腺癌或任何原因死亡的时间。其他疗效结果包括IDFS,包括第二原发性非乳腺癌,无病生存期(DFS)和总体生存期(OS)。
患者的人口统计学特征和基线肿瘤特征通常在治疗组之间保持平衡。中位年龄约为49岁(介于23-80岁之间),白人占73%,亚裔占9%,美洲印第安人或阿拉斯加土著人占6%,黑人或非洲裔美国人占3%。大多数患者(99.7%)是女性。各地区的入学情况如下:北美23%,欧洲54%,世界其他地区23%。肿瘤的预后特征包括激素受体状态(阳性:72%,阴性:28%),就诊时的临床阶段(无法操作:25%,可操作:75%)和术前治疗后的病理淋巴结状态(淋巴结阳性:46%,淋巴结阴性)或未评估:54%)在研究组之间相似。
大多数患者(77%)接受了含蒽环类药物的新辅助化疗方案。除曲妥珠单抗外,有20%的患者接受了另一种以HER2靶向的药物作为新辅助治疗的一部分。这些患者中有94%接受了帕妥珠单抗治疗。
中位随访40个月后,与曲妥珠单抗相比,接受KADCYLA的患者的IDFS统计学上显着改善。OS数据在IDFS分析时尚未成熟(1486例患者中有98例死亡[6.6%])。表8和图3总结了KATHERINE的功效结果。
一致的结果与观察到KADCYLA在IDFS方面跨基于分层因素,关键基线人口统计学和疾病特征,和在先治疗亚组。
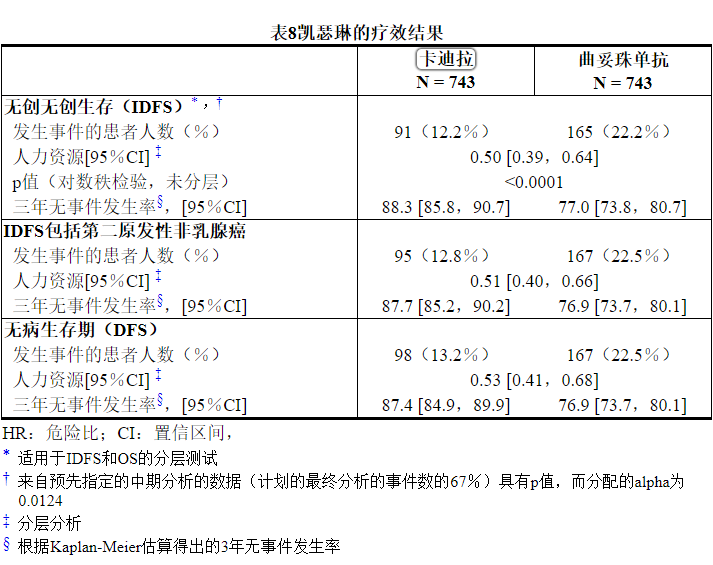
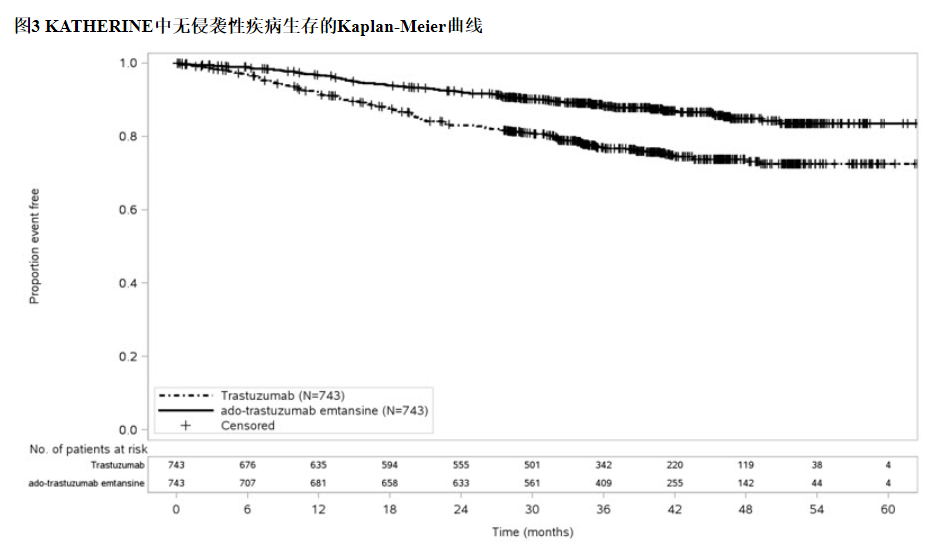
15参考
1. OSHA危险药物。 OSHA。http://www.osha.gov/SLTC/hazardousdrugs/index.html
16供应/存储和处理方式
16.1供应/存储方式
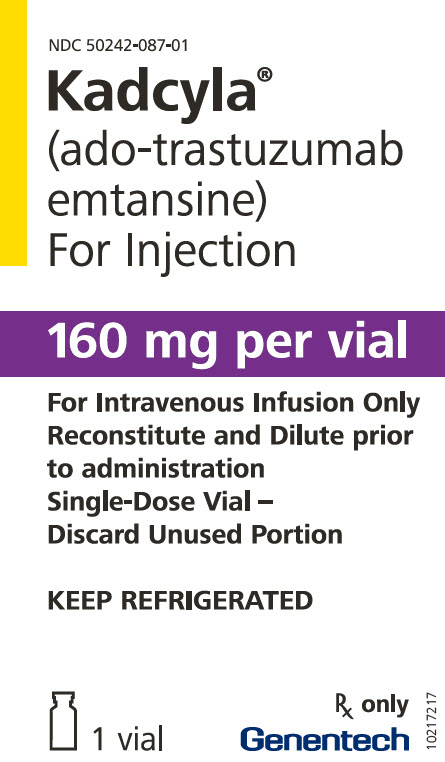
KADCYLA(阿托曲妥珠单抗丹参碱)的提供方式为:
将小瓶存放在2°C至8°C(36°F至46°F)的冰箱中,直到重新配制为止。 请勿冻结或摇动。
16.2特殊处理
遵循正确处理和处置抗癌药物的程序。
17患者咨询信息
肝毒性
◎告知患者严重肝损伤的可能性,并建议患者如果遇到急性肝炎的症状,例如恶心,呕吐,腹痛(特别是RUQ腹痛),黄疸,尿黑,全身瘙痒,厌食等,请立即就医。 。[见警告和注意事项(5.1) ]。
左心室功能不全
◎建议患者就下列任何情况立即联系医疗保健专业人员:新发或呼吸急促,咳嗽,脚踝/腿肿胀,心,24小时内体重增加超过5磅,头晕或失去知觉[参见警告和注意事项(5.2) ] 。
胚胎-胎儿毒性
◎建议孕妇和有生殖能力的女性在怀孕期间或在怀孕前七个月内接触KADCYLA会导致胎儿伤害。建议女性患者联系他们的卫生保健提供者与已知的或怀疑怀孕[见特殊人群中使用(8.1,8.3) ]。
◎建议孕妇在怀孕期间接触KADCYLA或在最后一次服用KADCYLA后7个月内怀孕的妇女,应制定监测怀孕结局的怀孕药物警戒计划。鼓励这些患者向Genentech报告其妊娠情况(请参阅“ 在特定人群中使用(8.1) ”)。
◎繁殖潜力的指教女性治疗期间和7个月内使用有效避孕的最后剂量后KADCYLA [见特殊人群中使用(8.1,8.3) ]。
◎建议具有生殖潜能的女性伴侣的男性患者在治疗期间以及最后一次使用 KADCYLA后 4个月内使用有效的避孕药[请参见在特定人群中使用(8.3) ]。
哺乳期
◎劝告妇女在治疗期间以及最后一次服用KADCYLA 后7个月内不要进行母乳喂养[见特定人群的使用(8.2) ]。
KADCYLA ® [费周折-曲妥单抗emtansine]
由:
Genentech,Inc制造。
罗氏集团成员
1 DNA Way
South San Francisco,CA 94080-4990
美国许可证号:1048KADCYLA是Genentech,Inc.的商标。
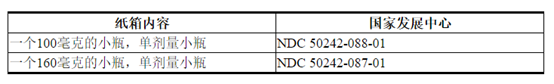
© 2019 Genentech,Inc.。标签的代表性示例(有关完整列表,请参见“ 如何提供”部分):
主要显示屏-100毫克瓶装纸箱
NDC 50242-088-01
Kadcyla ®
(ADO-曲妥珠单抗
emtansine)
对于注射每瓶100毫克
对于静脉输注,仅可
在
给药前重新配制并稀释
单剂量小瓶–
丢弃未使用的部分保持冷藏
仅Rx
1个小瓶
基因技术
10217212

主要显示屏-160毫克瓶装纸箱
NDC 50242-087-01
Kadcyla ®
(ADO-曲妥珠单抗
emtansine)
对于注射每瓶160毫克
对于静脉输注,仅可
在
给药前重新配制并稀释
单剂量小瓶–
丢弃未使用的部分保持冷藏
仅Rx
1个小瓶
基因技术
10217217
【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
本说明书来源于:美国FDA
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书。
FULL PRESCRIBING INFORMATION
WARNING: HEPATOTOXICITY, CARDIAC TOXICITY, EMBRYO-FETAL TOXICITY
Hepatotoxicity: Serious hepatotoxicity has been reported, including liver failure and death in patients treated with KADCYLA. Monitor serum transaminases and bilirubin prior to initiation of KADCYLA treatment and prior to each KADCYLA dose. Reduce dose or discontinue KADCYLA as appropriate in cases of increased serum transaminases or total bilirubin. (2.3, 5.1)
Cardiac Toxicity: KADCYLA administration may lead to reductions in left ventricular ejection fraction (LVEF). Evaluate left ventricular function in all patients prior to and during treatment with KADCYLA. Withhold treatment for clinically significant decrease in left ventricular function. (2.3, 5.2)
Embryo-Fetal Toxicity: Exposure to KADCYLA during pregnancy can result in embryo-fetal harm. Advise patients of these risks and the need for effective contraception. (5.3, 8.1, 8.3)
1 INDICATIONS AND USAGE
1.1 Metastatic Breast Cancer (MBC)
KADCYLA®, as a single agent, is indicated for the treatment of patients with HER2-positive, metastatic breast cancer who previously received trastuzumab and a taxane, separately or in combination. Patients should have either:
◎Received prior therapy for metastatic disease, or
◎Developed disease recurrence during or within six months of completing adjuvant therapy.
Select patients for therapy based on an FDA-approved companion diagnostic for KADCYLA [see Dosage and Administration (2.1)].
1.2 Early Breast Cancer (EBC)
KADCYLA, as a single agent, is indicated for the adjuvant treatment of patients with HER2-positive early breast cancer who have residual invasive disease after neoadjuvant taxane and trastuzumab -based treatment.
Select patients for therapy based on an FDA-approved companion diagnostic for KADCYLA [see Dosage and Administration (2.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients based on HER2 protein overexpression or HER2 gene amplification in tumor specimens [see Indications and Usage (1), Clinical Studies (14)]. Assessment of HER2 protein overexpression and/or HER2 gene amplification should be performed using FDA-approved tests specific for breast cancers by laboratories with demonstrated proficiency. Information on the FDA-approved tests for the detection of HER2 protein overexpression and HER2 gene amplification is available at: http://www.fda.gov/CompanionDiagnostics.
Improper assay performance, including use of sub-optimally fixed tissue, failure to utilize specified reagents, deviation from specific assay instructions, and failure to include appropriate controls for assay validation, can lead to unreliable results.
2.2 Recommended Doses and Schedules
Do not substitute trastuzumab for or with KADCYLA.
The recommended dose of KADCYLA is 3.6 mg/kg given as an intravenous infusion every 3 weeks (21-day cycle). Do not administer KADCYLA at doses greater than 3.6 mg/kg.
Closely monitor the infusion site for possible subcutaneous infiltration during drug administration [see Warnings and Precautions (5.9)].
First infusion: Administer infusion over 90 minutes. Observe patients during the infusion and for at least 90 minutes following the initial dose for fever, chills, or other infusion-related reactions [see Warnings and Precautions (5.5)].
Subsequent infusions: Administer over 30 minutes if prior infusions were well tolerated. Observe patients during the infusion and for at least 30 minutes after infusion.
Metastatic Breast Cancer (MBC)
Patients with MBC should receive treatment until disease progression or unmanageable toxicity.
Early Breast Cancer (EBC)
Patients with EBC should receive treatment for a total of 14 cycles unless there is disease recurrence or unmanageable toxicity.
2.3 Dose Modifications
Do not re-escalate the KADCYLA dose after a dose reduction is made.
If a planned dose is delayed or missed, administer as soon as possible; do not wait until the next planned cycle. Adjust the schedule of administration to maintain a 3-week interval between doses. Administer the infusion at the dose and rate the patient tolerated in the most recent infusion.
Slow or interrupt the infusion rate of KADCYLA if the patient develops an infusion-related reaction. Permanently discontinue KADCYLA for life-threatening infusion-related reactions [see Warnings and Precautions (5.5)].
Management of increased serum transaminases, hyperbilirubinemia, left ventricular dysfunction, thrombocytopenia, pulmonary toxicity or peripheral neuropathy may require temporary interruption, dose reduction or treatment discontinuation of KADCYLA as per guidelines provided in Tables 1 and 2.
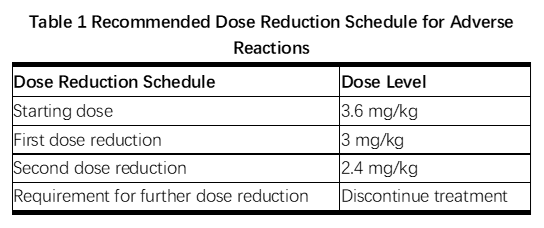
Table 2 Dose Modification Guidelines for KADCYLA
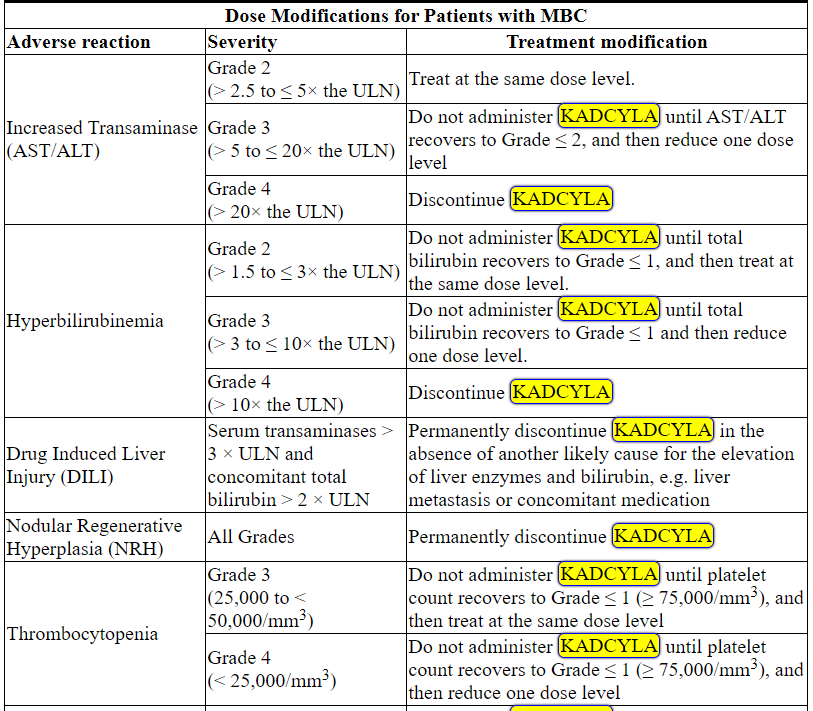
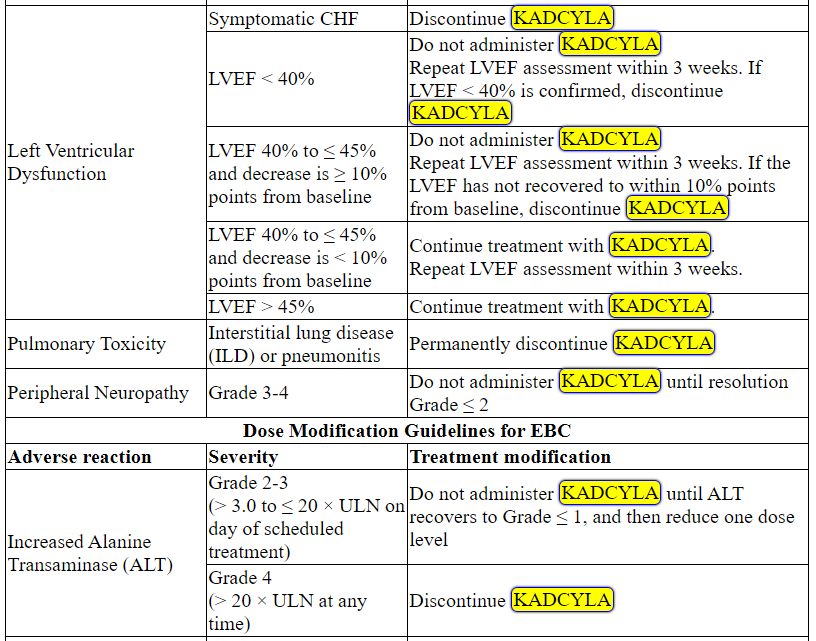
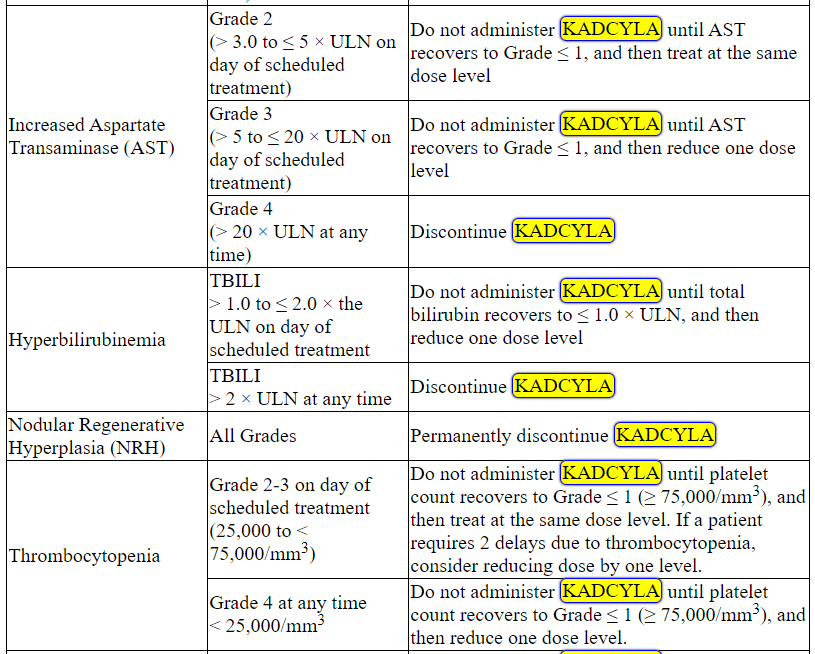
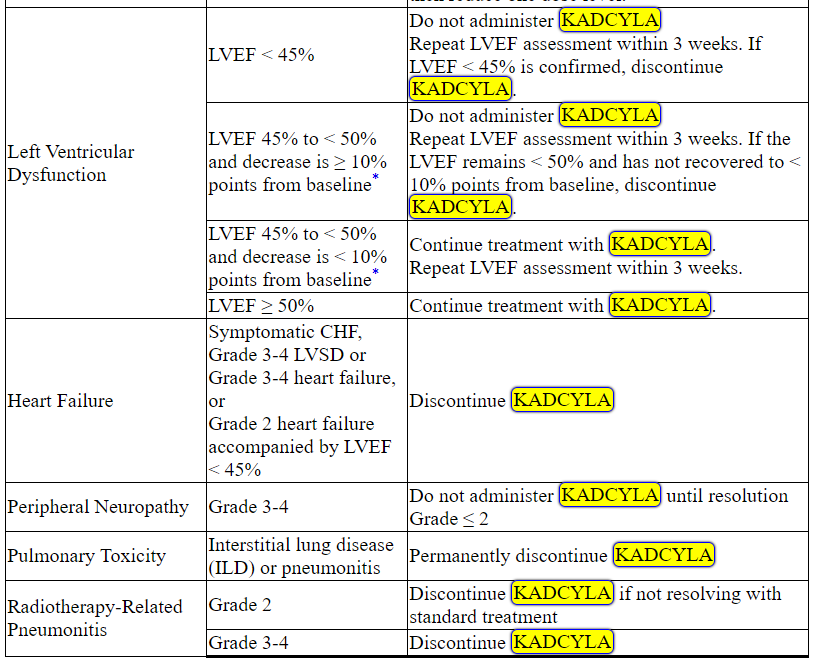
ALT = alanine transaminase; AST = aspartate transaminase, CHF = congestive heart failure, DILI = Drug Induced Liver Injury; LVEF = left ventricular ejection fraction, LVSD = left ventricular systolic dysfunction, TBILI = Total Bilirubin, ULN = upper limit of normal
* Prior to starting KADCYLA treatment
2.4 Preparation for Administration
In order to prevent medication errors it is important to check the vial labels to ensure that the drug being prepared and administered is KADCYLA (ado-trastuzumab emtansine) and not trastuzumab.
Administration:
◎Administer KADCYLA as an intravenous infusion only with a 0.2 or 0.22 micron in-line polyethersulfone (PES) filter. Do not administer as an intravenous push or bolus.
◎Do not mix KADCYLA, or administer as an infusion, with other medicinal products.
Reconstitution:
◎Use aseptic technique for reconstitution and preparation of dosing solution. Appropriate procedures for the preparation of chemotherapeutic drugs should be used.
◎Using a sterile syringe, slowly inject 5 mL of Sterile Water for Injection into the 100 mg KADCYLA vial, or 8 mL of Sterile Water for Injection into the 160 mg KADCYLA vial to yield a solution containing 20 mg/mL. Swirl the vial gently until completely dissolved. Do not shake. Inspect the reconstituted solution for particulates and discoloration.
◎The reconstituted solution should be clear to slightly opalescent and free of visible particulates. The color of the reconstituted solution should be colorless to pale brown. Do not use if the reconstituted solution contains visible particulates or is cloudy or discolored.
◎The reconstituted lyophilized vials should be used immediately following reconstitution with Sterile Water for Injection. If not used immediately, the reconstituted KADCYLA vials can be stored for up to 24 hours in a refrigerator at 2ºC to 8ºC (36°F to 46°F); discard unused KADCYLA after 24 hours. Do not freeze.
◎The reconstituted product contains no preservative and is intended for single-dose only.
Dilution:
Determine the correct dose (mg) of KADCYLA [see Dosage and Administration (2.1)].
◎Calculate the volume of the 20 mg/mL reconstituted KADCYLA solution needed.
◎Withdraw this amount from the vial and add it to an infusion bag containing 250 mL of 0.9% Sodium Chloride Injection. Do not use Dextrose (5%) solution.
◎Gently invert the bag to mix the solution in order to avoid foaming.
◎The diluted KADCYLA infusion solution should be used immediately. If not used immediately, the solution may be stored in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours prior to use. This storage time is additional to the time allowed for the reconstituted vials. Do not freeze or shake.
3 DOSAGE FORMS AND STRENGTHS
Lyophilized powder in single-dose vials: 100 mg per vial or 160 mg per vial of ado-trastuzumab emtansine.
4 CONTRAINDICATIONS
None.
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Hepatotoxicity, predominantly in the form of asymptomatic, transient increases in the concentrations of serum transaminases, has been observed in clinical trials with KADCYLA [see Adverse Reactions (6.1)]. Serious hepatotoxicity, including 3 fatal cases, has been observed in clinical trials (n=1624) with KADCYLA as single-agent. All fatal cases occurred in MBC clinical trials with KADCYLA, which included severe drug-induced liver injury and associated hepatic encephalopathy. Some of the patients experiencing hepatotoxicity had comorbidities and/or concomitant medications with known hepatotoxic potential.
Monitor serum transaminases and bilirubin prior to initiation of KADCYLA treatment and prior to each KADCYLA dose. Patients with known active liver disease (such as, hepatitis B virus or hepatitis C virus) were excluded from the EMILIA and KATHERINE studies [see Clinical Studies (14.1)]. Reduce the dose or discontinue KADCYLA as appropriate in cases of increased serum transaminases and/or total bilirubin [see Dosage and Administration (2.2)]. Permanently discontinue KADCYLA treatment in patients with serum transaminases > 3 × ULN and concomitant total bilirubin > 2 × ULN. KADCYLA has not been studied in patients with serum transaminases > 2.5 × ULN or bilirubin > 1.5 × ULN prior to the initiation of treatment.
In clinical trials of KADCYLA, cases of nodular regenerative hyperplasia (NRH) of the liver have been identified from liver biopsies (5 cases out of 1624 treated patients, one of which was fatal). Two of these five cases of NRH were observed in EMILIA and two were observed in KATHERINE [see Adverse Reactions (6.1)]. NRH is a rare liver condition characterized by widespread benign transformation of hepatic parenchyma into small regenerative nodules; NRH may lead to non-cirrhotic portal hypertension. The diagnosis of NRH can be confirmed only by histopathology. NRH should be considered in all patients with clinical symptoms of portal hypertension and/or cirrhosis-like pattern seen on the computed tomography (CT) scan of the liver but with normal transaminases and no other manifestations of cirrhosis. Upon diagnosis of NRH, KADCYLA treatment must be permanently discontinued.
5.2 Left Ventricular Dysfunction
Patients treated with KADCYLA are at increased risk of developing left ventricular dysfunction. A decrease of LVEF to < 40% has been observed in patients treated with KADCYLA. Serious cases of heart failure, with no fatal cases, have been observed in clinical trials with KADCYLA. In EMILIA, left ventricular dysfunction occurred in 1.8% of patients in the KADCYLA-treated group and 3.3% of patients in the lapatinib plus capecitabine-treated group. In KATHERINE, left ventricular dysfunction occurred in 0.4% of patients in the KADCYLA-treated group and 0.6% of patients in the trastuzumab-treated group [see Adverse Reactions (6.1)].
Assess LVEF prior to initiation of KADCYLA and at regular intervals (e.g. every three months) during treatment to ensure the LVEF is within the institution's normal limits. Treatment with KADCYLA has not been studied in patients with LVEF < 50% prior to initiation of treatment.
For patients with MBC, if, at routine monitoring, LVEF is < 40%, or is 40% to 45% with a 10% or greater absolute decrease below the pretreatment value, withhold KADCYLA and repeat LVEF assessment within approximately 3 weeks. Permanently discontinue KADCYLA if the LVEF has not improved or has declined further.
For patients with EBC, if, at routine monitoring, LVEF is < 45%, or is 45% to 49% with a 10% or greater absolute decrease below the pretreatment value, withhold KADCYLA and repeat LVEF assessment within approximately 3 weeks. Permanently discontinue KADCYLA if the LVEF has not improved or has declined further [see Dosage and Administration (2.2)].
Patients with a history of symptomatic congestive heart failure (CHF), serious cardiac arrhythmia, or history of myocardial infarction or unstable angina within 6 months were excluded from the EMILIA and KATHERINE studies [see Clinical Studies (14.1)].
5.3 Embryo-Fetal Toxicity
KADCYLA can cause fetal harm when administered to a pregnant woman. Cases of oligohydramnios, and oligohydramnios sequence manifesting as pulmonary hypoplasia, skeletal abnormalities and neonatal death were observed in the post-marketing setting in patients treated with trastuzumab, the antibody component of KADCYLA. DM1, the cytotoxic component of KADCYLA, can cause embryo-fetal toxicity based on its mechanism of action.
Verify the pregnancy status of females of reproductive potential prior to the initiation of KADCYLA. Advise pregnant women and females of reproductive potential that exposure to KADCYLA during pregnancy or within 7 months prior to conception can result in fetal harm. Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of KADCYLA [see Use in Specific Populations (8.1, 8.3)].
5.4 Pulmonary Toxicity
Cases of interstitial lung disease (ILD), including pneumonitis, some leading to acute respiratory distress syndrome or fatal outcome have been reported in clinical trials with KADCYLA. Signs and symptoms include dyspnea, cough, fatigue, and pulmonary infiltrates.
In patients with MBC, pneumonitis was reported at an incidence of 0.8% (7 out of 884 treated patients), with one case of Grade 3 pneumonitis. The overall incidence of pneumonitis was 1.2% in EMILIA. In KATHERINE, pneumonitis was reported at an incidence of 1.1% (8 out of 740 patients treated with KADCYLA), with one case of Grade 3 pneumonitis.
Radiation pneumonitis was reported at an incidence of 1.8% (11 out of 623 patients treated with adjuvant radiotherapy and KADCYLA), with 2 cases of Grade 3 radiation pneumonitis [see Adverse Reactions (6.1)].
Permanently discontinue treatment with KADCYLA in patients diagnosed with ILD or pneumonitis. For patients with radiation pneumonitis in the adjuvant setting, KADCYLA should be permanently discontinued for Grade ≥ 3 or for Grade 2 not responding to standard treatment [see Dose Modifications (2.2)].
Patients with dyspnea at rest due to complications of advanced malignancy, co-morbidities, and receiving concurrent pulmonary radiation therapy may be at increased risk of pulmonary toxicity.
5.5 Infusion-Related Reactions, Hypersensitivity Reactions
Treatment with KADCYLA has not been studied in patients who had trastuzumab permanently discontinued due to infusion-related reactions (IRRs) and/or hypersensitivity; treatment with KADCYLA is not recommended for these patients.
Infusion-related reactions, characterized by one or more of the following symptoms − flushing, chills, pyrexia, dyspnea, hypotension, wheezing, bronchospasm, and tachycardia have been reported in clinical trials of KADCYLA. In EMILIA, the overall incidence of IRRs in patients treated with KADCYLA was 1.4%. In KATHERINE, the overall incidence of IRRs in patients treated with KADCYLA was 1.6% [see Adverse Reactions (6.1)]. In most patients, these reactions resolved over the course of several hours to a day after the infusion was terminated. KADCYLA treatment should be interrupted in patients with severe IRR. KADCYLA treatment should be permanently discontinued in the event of a life-threatening IRR [see Dosage and Administration (2.2)]. Patients should be observed closely for IRR reactions, especially during the first infusion.
One case of a serious, allergic/anaphylactic-like reaction has been observed in clinical trials of single-agent KADCYLA. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use.
5.6 Hemorrhage
Cases of hemorrhagic events, including central nervous system, respiratory, and gastrointestinal hemorrhage, have been reported in clinical trials with KADCYLA. Some of these bleeding events resulted in fatal outcomes. In EMILIA, the overall incidence of hemorrhage was 32% in the KADCYLA-treated group and 16% in the lapatinib plus capecitabine-treated group. The incidence of Grade ≥ 3 hemorrhage was 1.8% in the KADCYLA-treated group and 0.8% in the lapatinib plus capecitabine-treated group. In KATHERINE, the overall incidence of hemorrhage was 29% in the KADCYLA-treated group and 10% in the trastuzumab-treated group. The incidence of Grade ≥ 3 hemorrhage was 0.4% in the KADCYLA-treated group, with one fatal case of intracranial hemorrhage, and 0.3% in the trastuzumab-treated group [see Adverse Reactions (6.1)]. Although, in some of the observed cases the patients were also receiving anti-coagulation therapy, antiplatelet therapy, or had thrombocytopenia, in others there were no known additional risk factors. Use caution with these agents and consider additional monitoring when concomitant use is medically necessary.
5.7 Thrombocytopenia
Thrombocytopenia, or decreased platelet count, was reported in clinical trials of KADCYLA (145 of 1624 treated patients with Grade ≥ 3; 494 of 1624 treated patients with any Grade). The majority of these patients had Grade 1 or 2 events (< LLN to ≥ 50,000/mm3) with the nadir occurring by day 8 and generally improving to Grade 0 or 1 (≥ 75,000/mm3) by the next scheduled dose. In clinical trials of KADCYLA, the incidence and severity of thrombocytopenia were higher in Asian patients.
In EMILIA, the overall incidence of thrombocytopenia was 31% in the KADCYLA-treated group and 3.3% in the lapatinib plus capecitabine-treated group [see Adverse Reactions (6.1)]. The incidence of Grade ≥ 3 thrombocytopenia was 15% in the KADCYLA-treated group and 0.4% in the lapatinib plus capecitabine-treated group. In Asian patients, the incidence of Grade ≥ 3 thrombocytopenia was 45% in the KADCYLA-treated group and 1.3% in the lapatinib plus capecitabine-treated group.
In KATHERINE, the overall incidence of thrombocytopenia was 29% in the KADCYLA-treated group and 2.4% in the trastuzumab-treated group [see Adverse Reactions (6.1)]. The incidence of Grade ≥ 3 thrombocytopenia was 6% in the KADCYLA-treated group and 0.3% in the trastuzumab-treated group. In Asian patients, the incidence of Grade ≥ 3 thrombocytopenia was 19% in the KADCYLA-treated group and 0% in the trastuzumab-treated group. The overall incidence of thrombocytopenia in the KADCYLA-treated group for Asian patients was 50%.
Monitor platelet counts prior to initiation of KADCYLA and prior to each KADCYLA dose [see Dosage and Administration (2.2)]. KADCYLA has not been studied in patients with platelet counts < 100,000/mm3 prior to initiation of treatment. In the event of decreased platelet count to Grade ≥ 3 (< 50,000/mm3) do not administer KADCYLA until platelet counts recover to Grade 1 (≥ 75,000/mm3) [see Dosage and Administration (2.2)]. Closely monitor patients with thrombocytopenia (< 100,000/mm3) and patients on anti-coagulant treatment during treatment with KADCYLA.
5.8 Neurotoxicity
Peripheral neuropathy, mainly as Grade 1 and predominantly sensory, was reported in clinical trials of KADCYLA (26 of 1624 treated patients with Grade ≥ 3; 435 of 1624 treated patients with any Grade). In EMILIA, the overall incidence of peripheral neuropathy was 21% in the KADCYLA-treated group and 14% in the lapatinib plus capecitabine-treated group [see Adverse Reactions (6.1)]. The incidence of Grade ≥ 3 peripheral neuropathy was 2.2% in the KADCYLA-treated group and 0.2% in the lapatinib plus capecitabine-treated group. In KATHERINE, the overall incidence of peripheral neuropathy was 32% in the KADCYLA-treated group and 17% in the trastuzumab-treated group. Peripheral neuropathy, including sensory and motor peripheral neuropathy, for KADCYLA treated patients 30% of cases were not resolved at the time of the primary IDFS analysis for KATHERINE. The incidence of Grade ≥ 3 peripheral neuropathy was 1.6% in the KADCYLA-treated group and 0.1% in the trastuzumab-treated group.
KADCYLA should be temporarily discontinued in patients experiencing Grade 3 or 4 peripheral neuropathy until resolution to Grade ≤ 2. Patients should be clinically monitored on an ongoing basis for signs or symptoms of neurotoxicity [see Nonclinical Toxicology (13.2)].
5.9 Extravasation
In KADCYLA clinical studies, reactions secondary to extravasation have been observed. These reactions, observed more frequently within 24 hours of infusion, were usually mild and comprised erythema, tenderness, skin irritation, pain, or swelling at the infusion site. Specific treatment for KADCYLA extravasation is unknown. The infusion site should be closely monitored for possible subcutaneous infiltration during drug administration.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
◎Hepatotoxicity [See Warnings and Precautions (5.1)]
◎Left Ventricular Dysfunction [See Warnings and Precautions (5.2)]
◎Embryo-Fetal Toxicity [See Warnings and Precautions (5.3)]
◎Pulmonary Toxicity [See Warnings and Precautions (5.4)]
◎Infusion-Related Reactions, Hypersensitivity Reactions [See Warnings and Precautions (5.5)]
◎Hemorrhage [See Warnings and Precautions (5.6)]
◎Thrombocytopenia [See Warnings and Precautions (5.7)]
◎Neurotoxicity [See Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the WARNINGS AND PRECAUTIONS reflect exposure to KADCYLA as a single agent at 3.6 mg/kg given as an intravenous infusion every 3 weeks (21-day cycle) in 1624 patients including 884 patients with HER2-positive metastatic breast cancer and 740 patients with HER2-positive early breast cancer (KATHERINE trial).
Metastatic Breast Cancer
In clinical trials, KADCYLA has been evaluated as single-agent in 884 patients with HER2-positive metastatic breast cancer. The most common (≥ 25%) adverse reactions were fatigue, nausea, musculoskeletal pain, hemorrhage, thrombocytopenia, headache, increased transaminases, constipation and epistaxis.
The adverse reactions described in Table 3 were identified in patients with HER2-positive metastatic breast cancer treated in the EMILIA trial [see Clinical Studies (14.1)]. Patients were randomized to receive KADCYLA or lapatinib plus capecitabine. The median duration of study treatment was 7.6 months for patients in the KADCYLA-treated group and 5.5 months and 5.3 months for patients treated with lapatinib and capecitabine, respectively.
In the EMILIA trial, 43% of patients experienced Grade ≥ 3 adverse reactions in the KADCYLA-treated group compared with 59% of patients in the lapatinib plus capecitabine-treated group.
Dose adjustments for KADCYLA were permitted [see Dosage and Administration (2.2)]. Thirty-two patients (7%) discontinued KADCYLA due to an adverse reaction, compared with 41 patients (8%) who discontinued lapatinib, and 51 patients (10%) who discontinued capecitabine due to an adverse reaction. The most common adverse reactions leading to KADCYLA discontinuation were thrombocytopenia and increased transaminases. Eighty patients (16%) treated with KADCYLA had adverse reactions leading to dose reductions. The most frequent adverse reactions leading to dose reduction of KADCYLA (in ≥ 1% of patients) included thrombocytopenia, increased transaminases, and peripheral neuropathy. Adverse reactions that led to dose delays occurred in 116 (24%) of KADCYLA treated patients. The most frequent adverse reactions leading to a dose delay of KADCYLA (in ≥ 1% of patients) were neutropenia, thrombocytopenia, leukopenia, fatigue, increased transaminases and pyrexia.
Table 3 reports the adverse reactions that occurred in patients in the KADCYLA-treated group (n=490) of the EMILIA trial. Selected laboratory abnormalities are shown in Table 4. The most common adverse reactions seen with KADCYLA in the randomized trial (frequency > 25%) were nausea, fatigue, musculoskeletal pain, hemorrhage, thrombocytopenia, increased transaminases, headache, and constipation. The most common NCI–CTCAE (version 3) Grade ≥ 3 adverse reactions (frequency > 2%) were thrombocytopenia, increased transaminases, anemia, hypokalemia, peripheral neuropathy and fatigue.
Table 3 Adverse Reactions Occurring in ≥ 10% of Patients on the KADCYLA Treatment Arm in the EMILIA Trial*
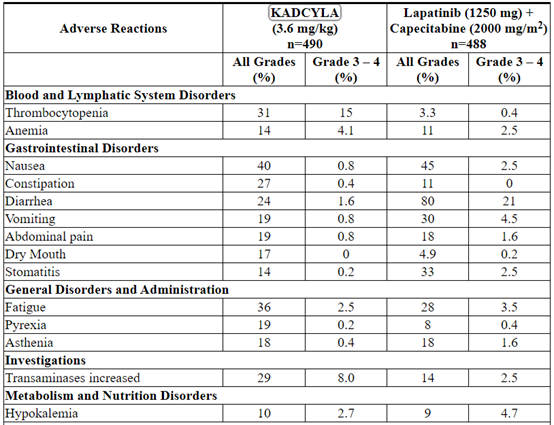
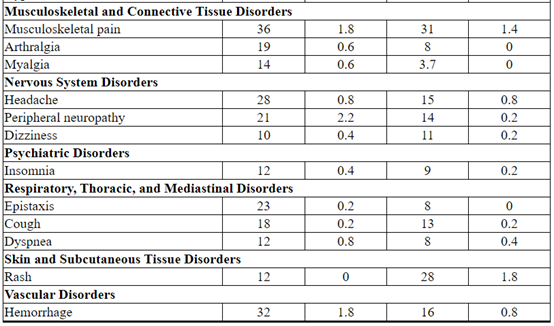

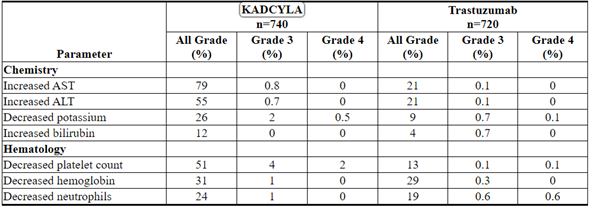
The following clinically relevant adverse reactions were reported in < 10% of patients in the KADCYLA-treated group in EMILIA: dyspepsia (9%), urinary tract infection (9%), chills (8%), dysgeusia (8%), neutropenia (7%), peripheral edema (7%), pruritus (6%), hypertension (5%), blood alkaline phosphatase increased (4.7%), vision blurred (4.5%), conjunctivitis (3.9%), dry eye (3.9%), lacrimation increased (3.3%), drug hypersensitivity (2.2%), left ventricular dysfunction (1.8%), infusion-related reaction (1.4%), pneumonitis (1.2%), nodular regenerative hyperplasia (0.4%), portal hypertension (0.4%).
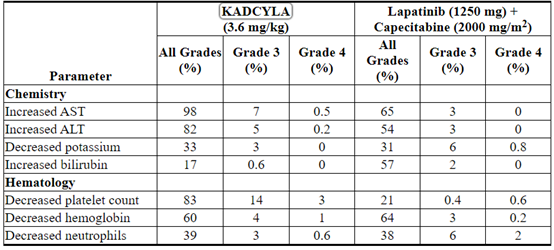
Table 4 Selected Laboratory Abnormalities (EMILIA)
Early Breast Cancer
KADCYLA has been evaluated as a single-agent in 740 patients with HER2-positive early breast cancer.
The adverse reactions described in Table 5 were identified in patients with HER2-positive early breast cancer treated in the KATHERINE trial [see Clinical Studies (14.2)]. Patients were randomized to receive KADCYLA or trastuzumab. The median duration of study treatment was 10 months for patients in the KADCYLA-treated group and 10 months for patients treated with trastuzumab.
One hundred and ninety (26%) patients experienced Grade ≥ 3 adverse reactions in the KADCYLA-treated group compared with 111 (15%) patients in the trastuzumab group. One hundred and thirty-three patients (18%) discontinued KADCYLA due to an adverse reaction, compared with 15 patients (2.1%) who discontinued trastuzumab due to an adverse reaction.
The most common adverse reactions leading to KADCYLA discontinuation (in ≥ 1% of patients) were platelet count decreased, blood bilirubin increased, ejection fraction decreased, AST increased, ALT increased, and peripheral neuropathy.
Dose adjustments for KADCYLA were permitted [see Dosage and Administration (2.2)]. One hundred and six patients (14%) treated with KADCYLA had dose reductions. The most frequent adverse reactions leading to dose reduction of KADCYLA (in ≥ 1% of patients) included thrombocytopenia, increased transaminases, blood bilirubin and fatigue. Adverse reactions that led to dose delays occurred in 106 (14%) of KADCYLA treated patients. The most frequent adverse reactions leading to a dose delay of KADCYLA (in ≥ 1% of patients) were neutropenia, thrombocytopenia and AST increased.
Selected laboratory abnormalities are shown in Table 6. The most common adverse reactions seen with KADCYLA in the randomized trial (frequency > 25%) were fatigue, nausea, increased transaminases, musculoskeletal pain, hemorrhage, thrombocytopenia, headache, peripheral neuropathy, and arthralgia. The most common NCI–CTCAE (version 3) Grade ≥ 3 adverse reactions (> 2%) were thrombocytopenia and hypertension.
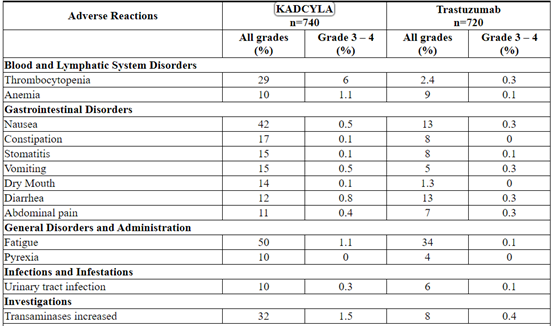
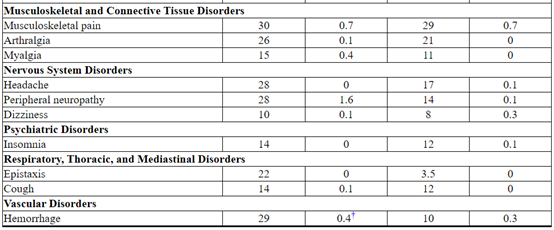
Table 5 Adverse Reactions Occurring in ≥ 10% of Patients in the KATHERINE Trial*
SMQ=standardized MedDRA queries
* Grouped terms were used for the following Adverse Reactions:
Thrombocytopenia: thrombocytopenia, platelet count decreased
Anemia: anemia, hemoglobin decreased
Stomatitis: stomatitis, mucosal inflammation, oropharyngeal pain
Abdominal pain: abdominal pain, abdominal pain upper
Urinary Tract Infection: urinary tract infection, cystitis
Transaminases Increased: transaminases increased, aspartate aminotransferase increased, alanine aminotransferase increased, gamma-glutamyltransferase increased, liver function test abnormal, hepatic enzyme increased, hepatic function abnormal
Musculoskeletal Pain: muscle spasms, musculoskeletal discomfort, musculoskeletal chest pain, back pain, pain in extremity, bone pain, musculoskeletal pain
Peripheral neuropathy: neuropathy peripheral, peripheral sensory neuropathy, peripheral motor neuropathy, paresthesia
Hemorrhage: Hemorrhage terms (excl laboratory terms) (SMQ, wide), Hemorrhage laboratory terms (SMQ, narrow)† Included one fatal hemorrhage.
The following clinically relevant adverse reactions were reported in < 10% of patients in the KADCYLA-treated group in KATHERINE: blood alkaline phosphatase increased (8%), dysgeusia (8%), dyspnea (8%), neutropenia (8%), blood bilirubin increased (7%), hypokalemia (7%), pruritus (7%), hypertension (6%), lacrimation increased (6%), chills (5%), dry eye (4.5%), dyspepsia (4.3%), peripheral edema (3.9%),vision blurred (3.9%), conjunctivitis (3.5%), left ventricular dysfunction (3.0%), drug hypersensitivity (2.7%), infusion-related reaction (1.6%), radiation pneumonitis (1.5%), pneumonitis (1.1%), rash (1.1%), asthenia (0.4%), nodular regenerative hyperplasia (0.3%).
Table 6 Selected Laboratory Abnormalities (KATHERINE)
6.2 Immunogenicity
As with all therapeutic proteins, there is the potential for an immune response to KADCYLA. A total of 1243 patients from seven clinical studies were tested at multiple time points for anti-drug antibody (ADA) responses to KADCYLA. Following KADCYLA dosing, 5.1% (63/1243) of patients tested positive for anti-KADCYLA antibodies at one or more post-dose time points. In clinical studies, 6.4% (24/376) of patients tested positive for anti-KADCYLA antibodies. In EMILIA, 5.2% (24/466) of patients tested positive for anti-KADCYLA antibodies, of which 13 were also positive for neutralizing antibodies. In KATHERINE, 3.7% (15/401) of patients tested positive for anti-KADCYLA antibodies, of which 5 were also positive for neutralizing antibodies. Due to the low incidence of ADA, conclusions cannot be made on the impact of anti-KADCYLA antibodies on the pharmacokinetics, safety, and efficacy of KADCYLA. The presence of KADCYLA in patient serum at the time of ADA sampling may interfere with the ability of this assay to detect anti-KADCYLA antibodies. As a result, data may not accurately reflect the true incidence of anti-KADCYLA antibody development.
Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication and the underlying disease. Therefore, comparison of the incidence of antibodies to KADCYLA with the incidence of antibodies to other products may be misleading. Clinical significance of anti-KADCYLA antibodies is not yet known.
6.3 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of KADCYLA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
◎Tumor lysis syndrome (TLS): Cases of possible TLS have been reported in patients treated with KADCYLA. Patients with significant tumor burden (e.g., bulky metastases) may be at a higher risk. Patients could present with hyperuricemia, hyperphosphatemia, and acute renal failure which may represent possible TLS. Providers should consider additional monitoring and/or treatment as clinically indicated.
7 DRUG INTERACTIONS
No formal drug-drug interaction studies with KADCYLA have been conducted. In vitro studies indicate that DM1, the cytotoxic component of KADCYLA, is metabolized mainly by CYP3A4 and to a lesser extent by CYP3A5. Concomitant use of strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole) with KADCYLA should be avoided due to the potential for an increase in DM1 exposure and toxicity. Consider an alternate medication with no or minimal potential to inhibit CYP3A4. If concomitant use of strong CYP3A4 inhibitors is unavoidable, consider delaying KADCYLA treatment until the strong CYP3A4 inhibitors have cleared from the circulation (approximately 3 elimination half-lives of the inhibitors) when possible. If a strong CYP3A4 inhibitor is coadministered and KADCYLA treatment cannot be delayed, patients should be closely monitored for adverse reactions.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Pharmacovigilance Program
There is a pregnancy pharmacovigilance program for KADCYLA. If KADCYLA is administered during pregnancy, or if a patient becomes pregnant while receiving KADCYLA or within 7 months following the last dose of KADCYLA, health care providers and patients should immediately report KADCYLA exposure to Genentech at 1-888-835-2555.
Risk Summary
KADCYLA can cause fetal harm when administered to a pregnant woman. There are no available data on the use of KADCYLA in pregnant women. Cases of oligohydramnios and oligohydramnios sequence manifesting as pulmonary hypoplasia, skeletal abnormalities, and neonatal death were observed in the postmarketing setting in patients treated with trastuzumab, the antibody component of KADCYLA [see Data]. Based on its mechanism of action, the DM1 component of KADCYLA can also cause embryo-fetal harm when administered to a pregnant woman [see Data]. Apprise the patient of the potential risks to a fetus. There are clinical considerations if KADCYLA is used in a pregnant woman, or if a patient becomes pregnant within 7 months following the last dose of KADCYLA [see Clinical Considerations].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Monitor women who received KADCYLA during pregnancy or within 7 months prior to conception for oligohydramnios. If oligohydramnios occurs, perform fetal testing that is appropriate for gestational age and consistent with community standards of care.
Data
Human Data
There are no available data on the use of KADCYLA in pregnant women. In the post-marketing setting, cases of oligohydramnios, and of oligohydramnios sequence, manifesting in the fetus as pulmonary hypoplasia, skeletal abnormalities and neonatal death were observed after treatment with trastuzumab during pregnancy. These case reports described oligohydramnios in pregnant women who received trastuzumab either alone or in combination with chemotherapy. In some case reports, amniotic fluid index increased after trastuzumab was stopped. In one case, trastuzumab therapy resumed after amniotic index improved, and oligohydramnios recurred.
Animal Data
There were no reproductive and developmental toxicology studies conducted with ado-trastuzumab emtansine. DM1, the cytotoxic component of KADCYLA, disrupts microtubule function. DM1 is toxic to rapidly dividing cells in animals and is genotoxic, suggesting it has the potential to cause embryotoxicity and teratogenicity. In studies where trastuzumab was administered to pregnant cynomolgus monkeys during the period of organogenesis at doses up to 25 mg/kg given twice weekly (about 7 times the clinical dose), trastuzumab crossed the placental barrier during the early (Gestation Days 20 to 50) and late (Gestation Days 120 to 150) phases of gestation. The resulting concentrations of trastuzumab in fetal serum and amniotic fluid were approximately 33% and 25%, respectively, of those present in the maternal serum but were not associated with adverse developmental effects.
8.2 Lactation
Risk Summary
There is no information regarding the presence of ado-trastuzumab emtansine in human milk, the effects on the breastfed infant, or the effects on milk production. DM1, the cytotoxic component of KADCYLA, may cause serious adverse reactions in breastfed infants based on its mechanism of action [see Data]. Advise women not to breastfeed during treatment and for 7 months following the last dose of KADCYLA.
Data
There were no animal lactation studies conducted with ado-trastuzumab emtansine or the cytotoxic component of KADCYLA (DM1). In lactating cynomolgus monkeys, trastuzumab was present in breast milk at about 0.3% of maternal serum concentrations after pre- (beginning Gestation Day 120) and post-partum (through Post-partum Day 28) doses of 25 mg/kg administered twice weekly (about 7 times the clinical dose of KADCYLA). Infant monkeys with detectable serum levels of trastuzumab did not exhibit any adverse effects on growth or development from birth to 1 month of age.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to the initiation of KADCYLA.
Contraception
Females
KADCYLA can cause embryo-fetal harm when administered during pregnancy. Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of KADCYLA [see Use in Specific Populations (8.1)].
Males
Because of the potential for genotoxicity, advise male patients with female partners of reproductive potential to use effective contraception during treatment with KADCYLA and for 4 months following the last dose.
Infertility
Based on results from animal toxicity studies, KADCYLA may impair fertility in females and males of reproductive potential. It is not known if the effects are reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of KADCYLA have not been established in pediatric patients.
8.5 Geriatric Use
Of the 495 patients who were randomized to KADCYLA in EMILIA [see Clinical Studies (14.1)], 65 patients (13%) were ≥ 65 years of age and 11 patients (2%) were ≥ 75 years of age. In patients ≥ 65 years old (n=138 across both treatment arms) the hazard ratios for progression-free survival (PFS) and overall survival (OS) were 1.06 (95% CI: 0.68, 1.66) and 1.05 (95% CI: 0.58, 1.91), respectively. No overall differences in the safety of KADCYLA were observed in patients aged ≥ 65 compared to patients < 65 years of age. EMILIA did not include sufficient numbers of patients aged ≥ 75 years to draw conclusions on the safety or effectiveness of KADCYLA in this age group.
Of the 743 patients who were randomized to KADCYLA in KATHERINE [see Clinical Studies (14.2)], 58 patients (8%) were ≥ 65 years of age and 2 patients (0.3%) were ≥ 75 years of age. No overall differences in the safety or effectiveness of KADCYLA were observed in patients aged ≥ 65 compared to patients < 65 years of age. KATHERINE did not include sufficient numbers of patients aged ≥ 75 years to draw conclusions on the safety or effectiveness of KADCYLA in this age group.
Population pharmacokinetic analysis indicates that age does not have a clinically meaningful effect on the pharmacokinetics of ado-trastuzumab emtansine [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dedicated renal impairment trial for KADCYLA has been conducted. Based on the population pharmacokinetics, as well as analysis of Grade 3 or greater adverse reactions and dose modifications, dose adjustments of KADCYLA are not needed in patients with mild (creatinine clearance [CLcr] 60 to 89 mL/min) or moderate (CLcr 30 to 59 mL/min) renal impairment. No dose adjustment can be recommended for patients with severe renal impairment (CLcr less than 30 mL/min) because of the limited data available [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No adjustment to the starting dose is required for patients with mild or moderate hepatic impairment [see Clinical Pharmacology (12.3)]. KADCYLA was not studied in patients with severe hepatic impairment. Closely monitor patients with hepatic impairment due to known hepatotoxicity observed with KADCYLA [see Warnings and Precautions, Hepatotoxicity (5.1)].
10 OVERDOSAGE
There is no known antidote for overdose of KADCYLA. In clinical trials, overdose of KADCYLA has been reported at approximately two times the recommended dose which resulted in Grade 2 thrombocytopenia (resolved 4 days later) and one death. In the fatal case, the patient incorrectly received KADCYLA at 6 mg/kg and died approximately 3 weeks following the overdose; a cause of death and a causal relationship to KADCYLA were not established.
11 DESCRIPTION
KADCYLA (ado-trastuzumab emtansine) is a HER2-targeted antibody-drug conjugate (ADC) which contains the humanized anti-HER2 IgG1, trastuzumab, covalently linked to the microtubule inhibitory drug DM1 (a maytansine derivative) via the stable thioether linker MCC (4-[N-maleimidomethyl] cyclohexane-1-carboxylate). Emtansine refers to the MCC-DM1 complex.
The antibody trastuzumab, is a well characterized recombinant monoclonal antibody product produced by mammalian (Chinese hamster ovary) cells, and the small molecule components (DM1 and MCC) are produced by chemical synthesis. Ado-trastuzumab emtansine contains an average of 3.5 DM1 molecules per antibody. Ado-trastuzumab emtansine has the following chemical structure:
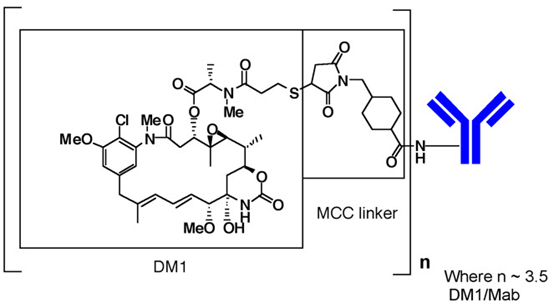
Note: The bracketed structure is DM1 plus MCC which represents the emtansine component. The n is, on average, 3.5 DM1 molecules per trastuzumab (Mab) molecule.
KADCYLA (ado-trastuzumab emtansine) is a sterile, white to off-white preservative free lyophilized powder in single-dose vials. Each vial contains 100 mg or 160 mg ado-trastuzumab emtansine. Following reconstitution, each single-dose vial contains ado-trastuzumab emtansine (20 mg/mL), polysorbate 20 [0.02% (w/v)], sodium succinate (10 mM), and sucrose [6% (w/v)] with a pH of 5.0. The resulting solution containing 20 mg/mL ado-trastuzumab emtansine is administered by intravenous infusion following dilution.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ado-trastuzumab emtansine is a HER2-targeted antibody-drug conjugate. The antibody is the humanized anti-HER2 IgG1, trastuzumab. The small molecule cytotoxin, DM1, is a microtubule inhibitor. Upon binding to sub-domain IV of the HER2 receptor, ado-trastuzumab emtansine undergoes receptor-mediated internalization and subsequent lysosomal degradation, resulting in intracellular release of DM1-containing cytotoxic catabolites. Binding of DM1 to tubulin disrupts microtubule networks in the cell, which results in cell cycle arrest and apoptotic cell death. In addition, in vitro studies have shown that similar to trastuzumab, ado-trastuzumab emtansine inhibits HER2 receptor signaling, mediates antibody-dependent cell-mediated cytotoxicity and inhibits shedding of the HER2 extracellular domain in human breast cancer cells that overexpress HER2.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of multiple doses of KADCYLA (3.6 mg/kg every 3 weeks) on the QTc interval was evaluated in an open label, single arm study in 51 patients with HER2-positive metastatic breast cancer. No large changes in the mean QT interval (i.e., > 20 ms) were detected in the study.
12.3 Pharmacokinetics
The pharmacokinetics of KADCYLA was evaluated in a phase 1 study and in a population pharmacokinetic analysis for the ado-trastuzumab emtansine conjugate (ADC) using pooled data from 5 trials in patients with breast cancer. A linear two-compartment model with first-order elimination from the central compartment adequately describes the ADC concentration-time profile. In addition to ADC, the pharmacokinetics of total antibody (conjugated and unconjugated trastuzumab), DM1 were also determined. The population pharmacokinetic analysis of ADC suggested no difference in KADCYLA exposure based on disease status (adjuvant vs. metastatic setting). The pharmacokinetics of KADCYLA are summarized below.
Distribution
Maximum concentrations (Cmax) of ADC and DM1 were observed close to the end of infusion. In EMILIA, mean (SD) ADC and DM1 Cycle 1 Cmax following KADCYLA administration was 83.4 (16.5) µg/mL and 4.61 (1.61) ng/mL, respectively. In KATHERINE, mean (SD) ADC and DM1 Cycle 1 Cmax following KADCYLA administration was 72.6 (24.3) µg/mL and 4.71 (2.25) ng/mL, respectively.
In vitro, the mean binding of DM1 to human plasma proteins was 93%. In vitro, DM1 was a substrate of P-glycoprotein (P-gp).
Based on population pharmacokinetic analysis, the central volume of distribution of ADC was 3.13 L.
Metabolism
In vitro studies indicate that DM1, the small molecule component of KADCYLA, undergoes metabolism by CYP3A4/5. DM1 did not inhibit or induce major CYP450 enzymes in vitro. In human plasma, ado-trastuzumab emtansine catabolites MCC-DM1, Lys-MCC-DM1, and DM1 were detected at low levels.
Elimination
Based on population pharmacokinetic analysis, following intravenous infusion of KADCYLA, the clearance of the ADC was 0.68 L/day and the elimination half-life (t1/2) was approximately 4 days. No accumulation of KADCYLA was observed after repeated dosing of intravenous infusion every 3 weeks.
Based on population pharmacokinetic analysis (n=671), body weight, sum of longest diameter of target lesions by RECIST, HER2 extracellular domain (ECD) concentrations, AST, albumin, and baseline trastuzumab concentrations were identified as statistically significant covariates for ado-trastuzumab emtansine clearance. However, the magnitude of effect of these covariates on ado-trastuzumab emtansine exposure suggests that, with the exception of body weight, these covariates are unlikely to have a clinically meaningful effect on KADCYLA exposure. Therefore, the body weight based dose of 3.6 mg/kg every 3 weeks without correction for other covariates is considered appropriate.
Effect of Renal Impairment
Based on population pharmacokinetic analysis in 668 patients, including moderate (CLcr 30 - 59 mL/min, n=53) and mild (CLcr 60 - 89 mL/min, n=254) renal impairment, indicate that pharmacokinetics of the ADC is not affected by mild to moderate renal impairment as compared to normal renal function (CLcr ≥ 90 mL/min, n=361). Data from only one patient with severe renal impairment (CLcr < 30 mL/min) is available [see Use in Specific Populations (8.7)].
Effect of Hepatic Impairment
The liver is a primary organ for eliminating DM1 and DM1-containing catabolites. The pharmacokinetics of ado-trastuzumab emtansine and DM1-containing catabolites were evaluated after the administration of 3.6 mg/kg of KADCYLA to metastatic HER2-positive breast cancer patients with normal hepatic function (n=10), mild (Child-Pugh A; n=10) and moderate (Child-Pugh B; n=8) hepatic impairment.
- Plasma concentrations of DM1 and DM1-containing catabolites (Lys-MCC-DM1 and MCC-DM1) were low and comparable between patients with and without hepatic impairment.
- Systemic exposures (AUC) of ado-trastuzumab emtansine at Cycle 1 in patients with mild and moderate hepatic impairment were approximately 38% and 67% lower than that of patients with normal hepatic function, respectively. Ado-trastuzumab emtansine exposure (AUC) at Cycle 3 after repeated dosing in patients with mild or moderate hepatic dysfunction was within the range observed in patients with normal hepatic function.
KADCYLA has not been studied in patients with severe hepatic impairment (Child-Pugh class C).
Effects of Age and Race
Based on population pharmacokinetic analysis, age (< 65 [n=577]; 65 - 75 (n=78); > 75 [n=16]) and race (Asian [n=73]; non-Asian [n=598]) do not have a clinically meaningful effect on the pharmacokinetics of ado-trastuzumab emtansine.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with ado-trastuzumab emtansine.
DM1 was aneugenic or clastogenic in an in vivo single-dose rat bone marrow micronucleus assay at exposures that were comparable to mean maximum concentrations of DM1 measured in humans administered KADCYLA. DM1 was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay.
Based on results from animal toxicity studies, KADCYLA may impair fertility in humans. In a single-dose toxicity study of ado-trastuzumab emtansine in rats, degeneration of seminiferous tubules with hemorrhage in the testes associated with increased weights of testes and epididymides at a severely toxic dose level (60 mg/kg; about 4 times the clinical exposure based on AUC) were observed. The same dose in female rats resulted in signs of hemorrhage and necrosis of the corpus luteum in ovaries. In monkeys dosed with ado-trastuzumab emtansine once every three weeks for 12 weeks (four doses), at up to 30 mg/kg (about 7 times the clinical exposure based on AUC), there were decreases in the weights of epididymides, prostate, testes, seminal vesicles and uterus, although the interpretation of these effects is unclear due to the varied sexual maturity of enrolled animals.
13.2 Animal Toxicology and/or Pharmacology
In monkeys, treatment with doses of ado-trastuzumab emtansine up to 30 mg/kg (about 7 times the clinical exposure based on AUC) caused dose dependent axonal degeneration in the sciatic nerve with hypertrophy or hyperplasia of the Schwann cells, and axonal degeneration of the dorsal funiculus in the spinal cord. Based on the mechanism of action of the cytotoxic component DM1, there is clinical potential for neurotoxicity [see Warnings and Precautions (5.8)].
14 CLINICAL STUDIES
14.1 Metastatic Breast Cancer
The efficacy of KADCYLA was evaluated in a randomized, multicenter, open-label trial (EMILIA) (NCT00829166) of 991 patients with HER2-positive, unresectable locally advanced or metastatic breast cancer. Prior taxane and trastuzumab-based therapy was required before trial enrollment. Patients with only prior adjuvant therapy were required to have disease recurrence during or within six months of completing adjuvant therapy. Breast tumor samples were required to show HER2 overexpression defined as 3+ IHC or FISH amplification ratio ≥ 2.0 determined at a central laboratory. Patients were randomly allocated (1:1) to receive lapatinib plus capecitabine or KADCYLA. Randomization was stratified by world region (United States, Western Europe, other), number of prior chemotherapy regimens for unresectable locally advanced or metastatic disease (0–1, > 1) and visceral versus non-visceral disease as determined by the investigators.
KADCYLA was given intravenously at 3.6 mg/kg on Day 1 of a 21-day cycle. Lapatinib was administered at 1250 mg/day orally once per day of a 21-day cycle and capecitabine was administered at 1000 mg/m2 orally twice daily on Days 1−14 of a 21-day cycle. Patients were treated with KADCYLA or lapatinib plus capecitabine until progression of disease, withdrawal of consent, or unacceptable toxicity. At the time of the primary analysis, median time on study drug was 5.7 months (range: 0–28.4) for KADCYLA, 4.9 months (range: 0–30.8) for lapatinib, and 4.8 months (range: 0–30.4) for capecitabine.
The co-primary efficacy outcomes of the study were progression-free survival (PFS) based on tumor response assessments by an independent review committee (IRC), and overall survival (OS). PFS was defined as the time from the date of randomization to the date of disease progression or death from any cause (whichever occurred earlier). Overall survival was defined as the time from the date of randomization to the date of death from any cause. Additional outcomes included PFS (based on investigator tumor response assessments), objective response rate (ORR), duration of response and time to symptom progression.
Patient demographics and baseline tumor characteristics were balanced between treatment arms. All patients had metastatic disease at study entry. The median age was approximately 53 years (range 24-84 years), 74% were White, 18% were Asian and 5% were Black. All but 5 patients were women. Twenty-seven percent of patients were enrolled in United States, 32% in Europe and 16% in Asia. Tumor prognostic characteristics including hormone receptor status (positive: 55%, negative: 43%), presence of visceral disease (68%) and non-visceral disease only (33%) and the number of metastatic sites (< 3: 61%, ≥ 3: 37%) were similar in the study arms.
The majority of patients (88%) had received prior systemic treatment in the metastatic setting. Twelve percent of patients had prior treatment only in the neoadjuvant or adjuvant setting and had disease relapse within 6 months of treatment. All but one patient received trastuzumab prior to study entry; approximately 85% of patients received prior trastuzumab in the metastatic setting. Over 99% percent of patients had received a taxane, and 61% of patients had received an anthracycline prior to study entry. Overall, patients received a median of 3 systemic agents in the metastatic setting. Among patients with hormone receptor-positive tumors, 44.4% received prior adjuvant hormonal therapy and 44.8% received hormonal therapy for locally advanced/metastatic disease.
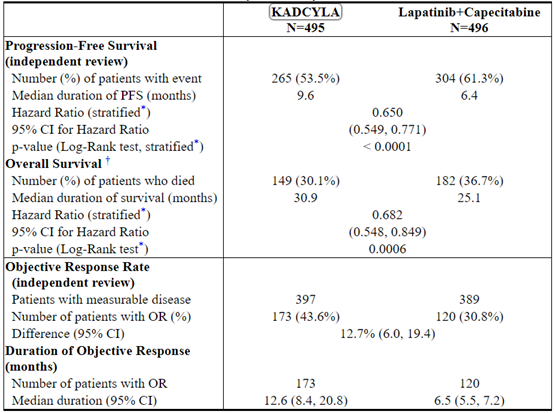
The randomized trial demonstrated a statistically significant improvement in IRC-assessed PFS in the KADCYLA-treated group compared with the lapatinib plus capecitabine-treated group [hazard ratio (HR) = 0.65, 95% CI: 0.55, 0.77, p < 0.0001], and an increase in median PFS of 3.2 months (median PFS of 9.6 months in the KADCYLA-treated group vs. 6.4 months in the lapatinib plus capecitabine group). See Table 7 and Figure 1. The results for investigator-assessed PFS were similar to those observed for IRC-assessed PFS.
At the time of PFS analysis, 223 patients had died. More deaths occurred in the lapatinib plus capecitabine arm (26%) compared with the KADCYLA arm (19%), however the results of this interim OS analysis did not meet the pre-specified stopping boundary for statistical significance. At the time of the second interim OS analysis, 331 events had occurred. The co-primary endpoint of OS was met; OS was significantly improved in patients receiving KADCYLA (HR = 0.68, 95% CI: 0.55, 0.85, p = 0.0006). This result crossed the pre-specified efficacy stopping boundary (HR = 0.73 or p = 0.0037). The median duration of survival was 30.9 months in the KADCYLA arm vs. 25.1 months in the lapatinib plus capecitabine arm. See Table 7 and Figure 2.
A treatment benefit with KADCYLA in terms of PFS and OS was observed in patient subgroups based on stratification factors, key baseline demographic and disease characteristics, and prior treatments. In the subgroup of patients with hormone receptor-negative disease (n=426), the hazard ratios for PFS and OS were 0.56 (95% CI: 0.44, 0.72) and 0.75 (95% CI: 0.54, 1.03), respectively. In the subgroup of patients with hormone receptor-positive disease (n=545), the hazard ratios for PFS and OS were 0.72 (95% CI: 0.58, 0.91) and 0.62 (95% CI: 0.46, 0.85), respectively. In the subgroup of patients with non-measurable disease (n=205), based on IRC assessments, the hazard ratios for PFS and OS were 0.91 (95% CI: 0.59, 1.42) and 0.96 (95% CI: 0.54, 1.68), respectively; in patients with measurable disease the hazard ratios were 0.62 (95% CI: 0.52, 0.75) and 0.65 (95% CI: 0.51, 0.82), respectively. The PFS and OS hazard ratios in patients who were younger than 65 years old (n=853) were 0.62 (95% CI: 0.52, 0.74) and 0.66 (95% CI: 0.52, 0.83), respectively. In patients ≥ 65 years old (n=138), the hazard ratios for PFS and OS were 1.06 (95% CI: 0.68, 1.66) and 1.05 (95% CI: 0.58, 1.91), respectively.
Table 7 Summary of Efficacy from EMILIA
PFS: progression-free survival; OR: objective response
* Stratified by world region (United States, Western Europe, other), number of prior chemotherapeutic regimens for locally advanced or metastatic disease (0-1 vs. > 1), and visceral vs. non-visceral disease.
† The second interim analysis for OS was conducted when 331 events were observed and the results are presented in this table.
Figure 1 Kaplan-Meier Curve of IRC-Assessed Progression-Free Survival for EMILIA
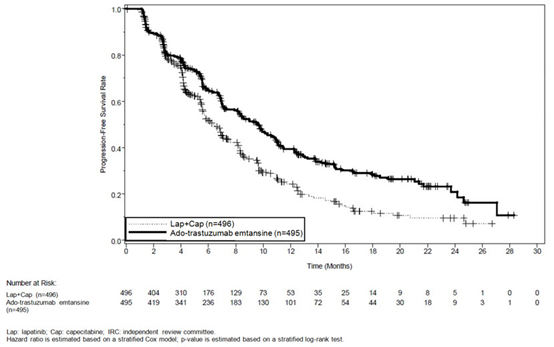
Figure 2 Kaplan-Meier Curve of Overall Survival for EMILIA
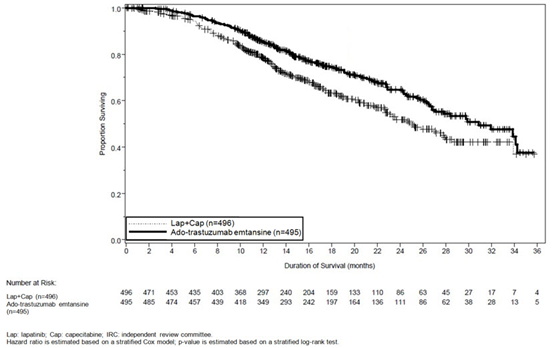
14.2 Early Breast Cancer
KATHERINE (NCT01772472) was a randomized, multicenter, open-label trial of 1486 patients with HER2-positive, early breast cancer. Patients were required to have had neoadjuvant taxane and trastuzumab-based therapy with residual invasive tumor in the breast and/or axillary lymph nodes. Patients received radiotherapy and/or hormonal therapy concurrent with study treatment as per local guidelines. Breast tumor samples were required to show HER2 overexpression defined as 3+ IHC or ISH amplification ratio ≥ 2.0 determined at a central laboratory using Ventana's PATHWAY anti-HER2-/neu (4B5) Rabbit Monoclonal Primary Antibody or INFORM HER2 Dual ISH DNA Probe Cocktail assays. Patients were randomized (1:1) to receive KADCYLA or trastuzumab. Randomization was stratified by clinical stage at presentation, hormone receptor status, preoperative HER2-directed therapy (trastuzumab, trastuzumab plus additional HER2-directed agent[s]), and pathological nodal status evaluation after preoperative therapy.
KADCYLA was given intravenously at 3.6 mg/kg on Day 1 of a 21-day cycle. Trastuzumab was given intravenously at 6 mg/kg on Day 1 of a 21-day cycle. Patients were treated with KADCYLA or trastuzumab for a total of 14 cycles unless there was recurrence of disease, withdrawal of consent, or unacceptable toxicity. At the time of the major efficacy outcome analysis, median treatment duration was 10 months for both KADCYLA- and trastuzumab-treated patients. Patients who discontinued KADCYLA for reasons other than disease recurrence could complete the remainder of the planned HER2-directed therapy with trastuzumab if appropriate based on toxicity considerations and investigator discretion.
The major efficacy outcome of the study was invasive disease-free survival (IDFS). IDFS was defined as the time from the date of randomization to first occurrence of ipsilateral invasive breast tumor recurrence, ipsilateral local or regional invasive breast cancer recurrence, distant recurrence, contralateral invasive breast cancer, or death from any cause. Additional efficacy outcomes included IDFS including second primary non-breast cancer, disease free survival (DFS), and overall survival (OS).
Patient demographics and baseline tumor characteristics were generally balanced between treatment arms. The median age was approximately 49 years (range 23-80 years), 73% were White, 9% were Asian, 6% were American Indian or Alaska Native and 3% were Black or African American. Most patients (99.7%) were women. Enrollment by region was as follows: 23% in North America, 54% in Europe and 23% throughout the rest of the world. Tumor prognostic characteristics including hormone receptor status (positive: 72%, negative: 28%), clinical stage at presentation (inoperable: 25%, operable: 75%) and pathological nodal status after preoperative therapy (node positive: 46%, node negative or not evaluated: 54%) were similar across study arms.
The majority of patients (77%) had received an anthracycline-containing neoadjuvant chemotherapy regimen. Twenty percent of patients received another HER2-targeted agent in addition to trastuzumab as a component of neoadjuvant therapy; 94% of these patients received pertuzumab.
After a median follow-up of 40 months, a statistically significant improvement in IDFS was observed in patients who received KADCYLA compared with trastuzumab. The OS data were not mature at the time of the IDFS analysis (98 deaths [6.6%] occurred in 1486 patients). The efficacy results from KATHERINE are summarized in Table 8 and Figure 3.
Consistent results were observed with KADCYLA in terms of IDFS across subgroups based on stratification factors, key baseline demographic and disease characteristics, and prior treatments.
Table 8 Efficacy Results from KATHERINE
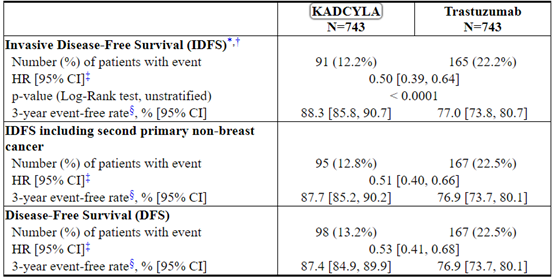
HR: Hazard Ratio; CI: Confidence Intervals,
* Hierarchical testing applied for IDFS and OS
† Data from the pre-specified interim analysis (67% of the number of events for the planned final analysis) with the p-value compared with the allocated alpha of 0.0124
‡ Unstratified analysis
§ 3-year event-free rate derived from Kaplan-Meier estimates
Figure 3 Kaplan-Meier Curve of Invasive Disease-Free Survival in KATHERINE
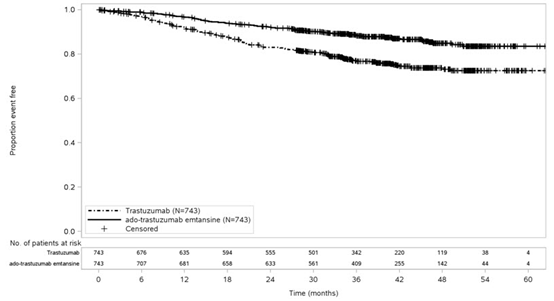
15 REFERENCES
1. OSHA Hazardous Drugs. OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied/Storage
KADCYLA (ado-trastuzumab emtansine) is supplied as:
Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F) until time of reconstitution. Do not freeze or shake.
16.2 Special Handling
Follow procedures for proper handling and disposal of anticancer drugs.
17 PATIENT COUNSELING INFORMATION
Hepatotoxicity
◎Inform patients of the possibility of severe liver injury and advise patients to immediately seek medical attention if they experience symptoms of acute hepatitis such as nausea, vomiting, abdominal pain (especially RUQ abdominal pain), jaundice, dark urine, generalized pruritus, anorexia, etc. [see Warnings and Precautions (5.1)].
Left Ventricular Dysfunction
◎Advise patients to contact a health care professional immediately for any of the following: new onset or worsening shortness of breath, cough, swelling of the ankles/legs, palpitations, weight gain of more than 5 pounds in 24 hours, dizziness or loss of consciousness [see Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
◎Advise pregnant women and females of reproductive potential that KADCYLA exposure during pregnancy or within 7 months prior to conception can result in fetal harm. Advise female patients to contact their healthcare provider with a known or suspected pregnancy [see Use in Specific Populations (8.1, 8.3)].
◎Advise women who are exposed to KADCYLA during pregnancy or who become pregnant within 7 months following the last dose of KADCYLA that there is a pregnancy pharmacovigilance program that monitors pregnancy outcomes. Encourage these patients to report their pregnancy to Genentech [see Use in Specific Populations (8.1)].
◎Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of KADCYLA [see Use in Specific Populations (8.1, 8.3)].
◎Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 4 months following the last dose of KADCYLA [see Use in Specific Populations (8.3)].
Lactation
◎Advise women not to breastfeed during treatment and for 7 months after the last dose of KADCYLA [see Use in Specific Populations (8.2)].
KADCYLA® [ado-trastuzumab emtansine]
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
U.S. License No: 1048KADCYLA is a trademark of Genentech, Inc.
©2019 Genentech, Inc.Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
PRINCIPAL DISPLAY PANEL - 100 mg Vial Carton
NDC 50242-088-01
Kadcyla®
(ado-trastuzumab
emtansine)
For Injection100 mg per vial
For Intravenous Infusion Only
Reconstitute and Dilute prior
to administration
Single-Dose Vial –
Discard Unused PortionKEEP REFRIGERATED
Rx only
1 vial
Genentech
10217212

PRINCIPAL DISPLAY PANEL - 160 mg Vial Carton
NDC 50242-087-01
Kadcyla®
(ado-trastuzumab
emtansine)
For Injection160 mg per vial
For Intravenous Infusion Only
Reconstitute and Dilute prior
to administration
Single-Dose Vial –
Discard Unused PortionKEEP REFRIGERATED
Rx only
1 vial
Genentech
10217217
【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。