商品详情
返回产品目录

商品包装及说明书因厂家更换频繁,如有不符以实物为主
康奈非尼胶囊
国际零售参考价:¥**/盒
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。
1适应症和用途
BRAFTOVI ®被指示时,在与组合binimetinib,用于患者的治疗不可切除或转移性黑色素瘤与BRAF V600E或V600K突变,由FDA批准的试验所检测[见剂量和给药方法(2.1) ]。
使用限制
未将BRAFTOVI指示用于治疗野生型BRAF黑色素瘤的患者[请参阅警告和注意事项(5.2) ]。
2用法用量
2.1患者选择
在启动BRAFTOVI之前,请确认肿瘤标本中存在BRAF V600E或V600K突变[见警告和注意事项(5.2),临床研究(14) ]。有关FDA批准的用于检测黑色素瘤中BRAF V600E和V600K突变的测试的信息,请访问:http://www.fda.gov/CompanionDiagnostics。
2.2推荐剂量
推荐的BRAFTOVI剂量为450 mg(六次75 mg胶囊),每天口服一次,与宾尼替尼联合使用,直至疾病进展或出现不可接受的毒性。有关推荐的比尼替尼剂量信息,请参阅比尼替尼处方信息。
BRAFTOVI可以与食物一起或不与食物一起服用[参见临床药理学(12.3) ]。不要在下一次服用BRAFTOVI后12小时内服用错过的BRAFTOVI。
如果在服用BRAFTOVI后出现呕吐,请不要再服用其他剂量,而要继续服用下一个预定的剂量。
2.3不良反应的剂量修改
如果拒绝使用宾尼替尼,则每天一次减少BRAFTOVI的最大剂量至300 mg,直到恢复宾尼替尼[请参阅警告和注意事项(5.7) ]。
表1列出了与BRAFTOVI相关的不良反应的剂量减少。
表1:减少BRAFTOVI不良反应的建议剂量

表2列出了与BRAFTOVI相关的不良反应的剂量调整方法。
表2:BRAFTOVI对不良反应的推荐剂量修改

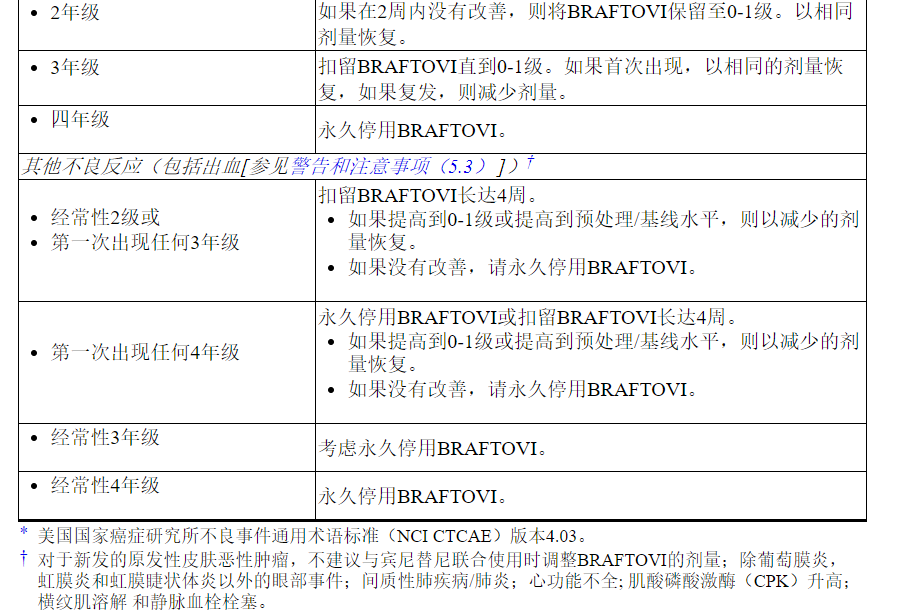
有关与Binimetinib相关的不良反应的剂量调整,请参考Binimetinib处方信息。
2.4与强或中度CYP3A4抑制剂共同给药的剂量修饰
在用BRAFTOVI治疗期间,避免与强效或中度CYP3A4抑制剂共同给药。如果不可避免地需要与强效或中度CYP3A4抑制剂合用,则根据表3中的建议降低BRAFTOVI剂量。在抑制剂停止3至5个消除半衰期后,恢复开始使用之前的BRAFTOVI剂量。 CYP3A4抑制剂[见药物相互作用(7.1),临床药理学(12.3) ]。
表3:与强或中度CYP3A4抑制剂共同给药的BRAFTOVI推荐的减量剂量
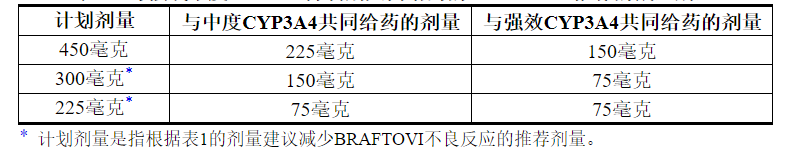
3剂型和强度
胶囊:75毫克,硬明胶,米色盖帽上的程式化“ A”,白色主体上的“ LGX 75毫克”
4禁忌症
没有。
5警告和注意事项
5.1新的原发性恶性肿瘤
在用BRAF抑制剂治疗的患者中已经观察到新的原发性恶性肿瘤,皮肤性和非皮肤性恶性肿瘤,并可能在BRAFTOVI中发生。
皮肤恶性肿瘤
在COLUMBUS中,接受BRAFTOVI联合比米替尼治疗的患者中,皮肤鳞状细胞癌(cuSCC)(包括角膜棘皮瘤(KA))发生率为2.6%,基底细胞癌发生率为1.6%。首次出现cuSCC / KA的中位时间为5.8个月(范围为1到9个月)[请参阅不良反应(6.1) ]。
对于接受BRAFTOVI单药治疗的患者,cuSCC / KA报告为8%,基底细胞癌为1%,新发原发性黑色素瘤为5%。
在开始治疗之前,治疗期间每2个月以及停药后最多6个月进行皮肤病学评估。通过切除和皮肤病理学评估处理可疑的皮肤病变。不建议对新发的原发性皮肤恶性肿瘤进行剂量调整。
非即时恶性肿瘤
基于其作用机制,BRAFTOVI可能通过突变或其他机制促进与RAS激活相关的恶性肿瘤[请参阅警告和注意事项(5.2) ]。监测接受BRAFTOVI治疗的患者的非皮肤恶性肿瘤的体征和症状。停止BRAFTOVI治疗RAS突变阳性的非皮肤恶性肿瘤[见剂量和给药方法(2.3) ]。
5.2 BRAF野生型肿瘤的肿瘤促进
体外实验已证明,暴露于BRAF抑制剂的BRAF野生型细胞中MAP激酶信号传导具有反常激活作用,并能增加细胞增殖。在启动BRAFTOVI之前,请确认BRAF V600E或V600K突变的证据[请参阅适应症和用法(1),剂量和用法(2.1) ]。
5.3出血
当BRAFTOVI与Binimetinib并用时,会发生出血。在哥伦布,接受BRAFTOVI联合比尼替尼治疗的患者中有19%发生出血;3.2%的患者发生3级以上出血。最常见的出血事件是胃肠道疾病,包括直肠出血(4.2%),便血(3.1%)和痔疮出血(1%)。在发生新的或进行性脑转移的情况下,致命的颅内出血发生在1.6%的患者中。
根据不良反应的严重程度,停药,减少剂量或永久中止[参见剂量和用法(2.3),不良反应(6.1) ]。
5.4葡萄膜炎
据报道,BRAFTOVI联合比尼替尼治疗的患者出现葡萄膜炎,包括虹膜炎和虹膜睫状体炎。在COLUMBUS中,接受BRAFTOVI和Binimetinib联合治疗的患者中葡萄膜炎的发生率为4%。
每次就诊时评估视觉症状。定期进行眼科评估,检查是否有新的或恶化的视觉障碍,并跟踪新的或持续的眼科检查结果。根据不良反应的严重程度,停药,减少剂量或永久中止[参见剂量和用法(2.3),不良反应(6.1) ]。
5.5 QT延长
在某些患者中,BRAFTOVI与剂量依赖性QTc间隔延长有关[见临床药理学(12.2) ]。在COLUMBUS中,接受BRAFTOVI联合比米替尼治疗的患者中有0.5%(1/192)的患者QTcF升高至> 500 ms。
监测已患有QTc延长或有发生QTc延长的重大风险的患者,包括已知的长时间QT综合征,临床上明显的心律失常,严重或不受控制的心力衰竭以及服用与QT延长相关的其他药物的患者。在服用BRAFTOVI之前和期间纠正低钾血症和低镁血症。如果QTc> 500 ms,请停药,降低剂量或永久停药[请参阅剂量和用法(2.3),不良反应(6.1) ]。
5.6胚胎-胎儿毒性
根据其作用机理,BRAFTOVI对孕妇给药可引起胎儿伤害。恩哥拉非尼在大鼠和兔子中产生胚胎-胎儿发育变化,并且在大于或等于剂量的情况下对兔子具有流产作用,所导致的暴露量是建议剂量下人暴露量的26倍(大鼠)和178倍(兔) 450 mg,低剂量时无明显发现。
建议妇女注意胎儿的潜在危险。繁殖潜力的指教雌性BRAFTOVI的最终剂量后使用有效的避孕,非激素的方法,因为BRAFTOVI可以呈现激素避孕药是无效的,在治疗过程中和2周[见特殊人群中使用(8.1,8.3) ]。
5.7与BRAFTOVI作为单一代理人相关的风险
与将BRAFTOVI与Binimetinib组合使用时相比,当BRAFTOVI作为单一药物使用时,会增加某些不良反应的风险。接受BRAFTOVI单药治疗的患者中有21%发生3或4级皮肤病反应,而接受BRAMETOVI合并比尼替尼治疗的患者发生2%[见警告和注意事项(5.1),不良反应(6.1) ]。
如果Binimetinib暂时中断或永久停用,请按照建议的剂量减少BRAFTOVI的剂量[参见剂量和用法(2.3) ]。
5.8联合治疗相关的风险
BRAFTOVI被指定与Binimetinib组合使用。有关适用于联合使用治疗的其他风险信息,请参考比尼替尼处方信息。
6不良反应
标签上其他地方描述了以下不良反应:
- 新的原发性恶性肿瘤[请参阅警告和注意事项(5.1) ]
- 出血[请参阅警告和注意事项(5.3) ]
- 葡萄膜炎[请参阅警告和注意事项(5.4) ]
- QT延长[请参阅警告和注意事项(5.5) ]
6.1临床试验经验
由于临床试验是在广泛不同的条件下进行的,因此无法将在某种药物的临床试验中观察到的不良反应率直接与另一种药物的临床试验中观察到的不良反应率进行比较,并且可能无法反映实际中观察到的不良反应率。
192例BRAF V600突变阳性的不可切除或转移性黑色素瘤患者接受BRAFTOVI(450 mg每天一次)联合binimetinib(45 mg每天两次)的随机开放标签有效患者中描述了BRAFTOVI联合比奈替尼的安全性对照试验(COLUMBUS)。
COLUMBUS试验[参见临床研究(14) ]排除了具有吉尔伯特综合症病史,左心室射血分数异常,QTc延长(> 480毫秒),高血压不受控制以及视网膜静脉阻塞的病史或当前证据的患者。接受BRAFTOVI联合比尼替尼治疗的患者的中位暴露时间为11.8个月,接受维拉非尼治疗的患者为6.2个月。
接受BRAFTOVI联合比尼替尼治疗的患者最常见的不良反应(≥25%)为疲劳,恶心,呕吐,腹痛和关节痛。
接受BRAFTOVI合并比尼替尼治疗的患者中有30%发生不良反应,导致BRAFTOVI剂量中断。最常见的是恶心(7%),呕吐(7%)和发热(4%)。接受BRAFTOVI合并比尼替尼治疗的患者中有14%发生不良反应,导致BRAFTOVI剂量降低;最常见的是关节痛(2%),疲劳(2%)和恶心(2%)。接受BRAFTOVI联合比尼替尼治疗的患者中有5%(5%)出现不良反应,导致BRAFTOVI永久停用;最常见的是2%的出血和1%的头痛。
表4和表5分别显示了在COLUMBUS中鉴定出的药物不良反应和实验室异常。COLUMBUS试验的目的不是表4所列任何具体不良反应,与vemurafenib相比,BRAFTOVI与Binimetinib联用的宾奈替尼不良反应率在统计学上有显着差异。
表4:在哥伦布中≥10%接受BRAFTOVI的患者与Binimetinib联合发生的不良反应*
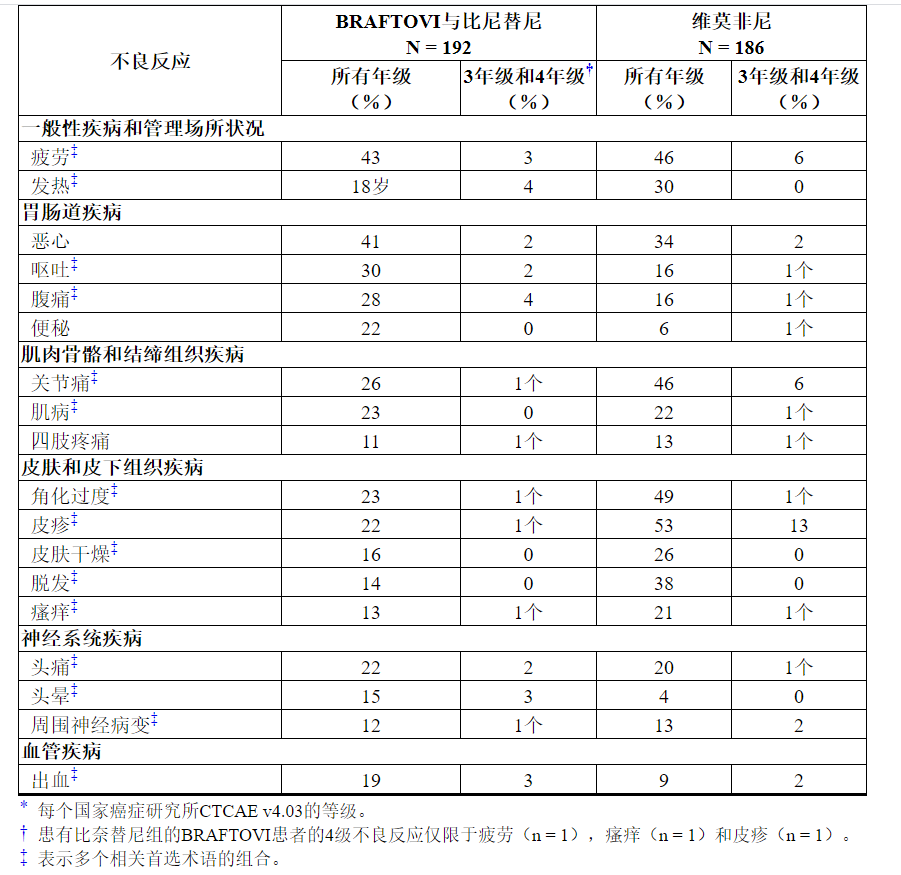
与BRAFTOVI联合比尼替尼联合使用时,BRAFTOVI单独使用会增加某些不良反应的风险。与单用BRAFTOVI合并比尼替尼的患者相比,每天一次口服BRAFTOVI 300 mg的患者与下列患者相比,观察到以下不良反应的发生率更高(≥5%):掌plant足部红斑感觉异常综合征(51%vs. 7) %),角化过度(57%对23%),皮肤干燥(38%对16%),红斑(16%对7%),皮疹(41%对22%),脱发(56%对22%) 14%),瘙痒(31%对13%),关节痛(44%对26%),肌病(33%对23%),背痛(15%对9%),味觉不良(13%对6%)和痤疮样皮炎(8%对3%)。
少于10%的接受BRAFTOVI联合比尼替尼治疗的患者发生的其他临床重要不良反应是:
神经系统疾病:面部轻瘫
胃肠道疾病:胰腺炎
皮肤和皮下组织疾病:脂膜炎
免疫系统疾病:药物超敏反应
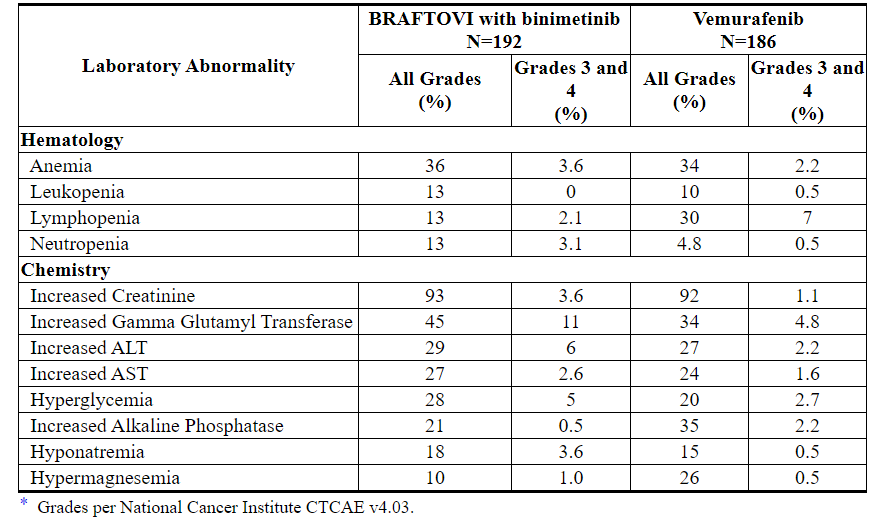
表5:在哥伦布中接受BRAFTOVI联合比尼替尼治疗的患者中≥10%(所有等级)的实验室异常*
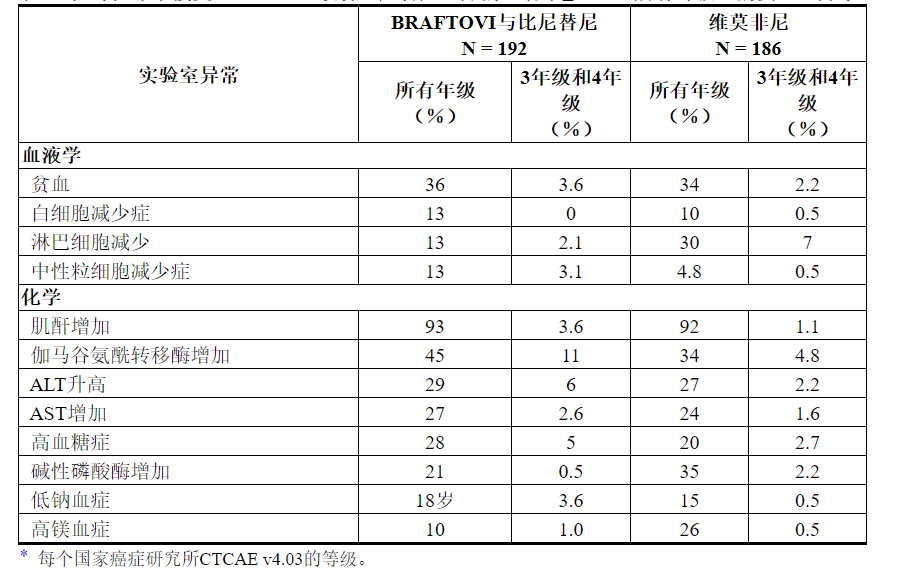
7药物相互作用
7.1其他药物对BRAFTOVI的影响
CYP3A4抑制剂
将BRAFTOVI与强或中度CYP3A4抑制剂同时给药会增加encorafenib的血药浓度,并可能增加encorafenib的不良反应[见临床药理学(12.3) ]。避免BRAFTOVI与强或中度CYP3A4抑制剂(包括葡萄柚汁)共同给药。如果无法避免同时使用强效或中效CYP3A4抑制剂,则按照推荐的剂量进行调整[见剂量和给药方法(2.4) ]。
强或中度CYP3A4诱导剂
将BRAFTOVI与强或中度CYP3A4诱导剂并用可能会降低encorafenib的血浆浓度并可能降低encorafenib的疗效[见临床药理学(12.3) ]。避免与BRAFTOVI并用强或中度CYP3A4诱导剂。
7.2 BRAFTOVI对其他药物的影响
敏感的CYP3A4底物
将BRAFTOVI与敏感的CYP3A4底物同时给药可能导致这些药物的毒性增加或疗效降低。
BRAFTOVI与激素避孕药(CYP3A4底物)的共同给药可导致浓度降低和激素避孕药效下降。避免激素避孕药[见在特定人群中使用(8.3) ]。
7.3延长QT间隔的药物
BRAFTOVI与剂量依赖性QTc间隔延长有关。避免将BRAFTOVI与具有延长QT / QTc间隔的已知潜力的药物合用[请参阅警告和注意事项(5.5),临床药理学(12.2) ]。
8在特定人群中的使用
8.1怀孕
风险摘要
根据其作用机理,BRAFTOVI可以在孕妇中引起胎儿伤害[见临床药理学(12.1) ]。没有关于怀孕期间使用BRAFTOVI的可用临床数据。在动物生殖研究中,encorafenib会在大鼠和兔子中产生胚胎-胎儿发育变化,并且在兔子身上流产,剂量大于或等于导致人类暴露量约26倍(大鼠)和178倍(兔子)的剂量。在450 mg的临床剂量下暴露,在较低剂量下无明确发现(参见数据)。建议孕妇注意胎儿的潜在危险。
在美国普通人群中,临床公认的怀孕中主要先天缺陷和流产的估计背景风险分别为2%至4%和15%至20%。
数据
动物资料
在生殖毒性研究中,在器官发生期间向大鼠施用恩可拉非尼会导致母体毒性,胎儿体重减少以及总骨骼变异的发生率增加,剂量为20 mg / kg /天(约为人类暴露量的26倍)。推荐的临床剂量为450 mg,每天一次,浓度-时间曲线[AUC]下的面积)。在怀孕的兔子中,施用恩可拉非尼在器官发生期间导致母体毒性,胎儿体重下降,总骨骼变异发生率增加以及植入后损失增加,包括剂量为75 mg / kg / day的妊娠完全丧失(约为人类暴露量的178倍)根据AUC,建议的临床剂量为450 mg,每天一次)。虽然尚未进行正式的胎盘移植研究,但大鼠和家兔在胎儿血浆中的encorafenib暴露分别高达母体暴露的1.7%和0.8%。
8.2哺乳
风险摘要
没有关于母乳中encorafenib或其代谢产物的存在或encorafenib对母乳喂养婴儿或产奶量的影响的数据。由于BRAFTOVI对母乳喂养的婴儿可能产生严重的不良反应,因此建议妇女在BRAFTOVI治疗期间和最终剂量后的两周内不要母乳喂养。
8.3生殖潜力的男性和女性
验孕
在开始BRAFTOVI之前,请验证具有生殖潜能的女性的怀孕状况[请参见在特定人群中使用(8.1) ]。
避孕
当BRAFTOVI给予孕妇时,可能会造成胎儿伤害[见在特定人群中使用(8.1) ]。
女性
建议有生育潜力的女性在用BRAFTOVI治疗期间和最终剂量后的2周内使用有效的避孕方法。建议患者使用非激素避孕方法,因为BRAFTOVI有可能使激素避孕药无效[见药物相互作用(7.2) ]。
不孕症
雄性
根据在450 mg临床剂量下人暴露量约13倍的雄性大鼠中的发现,BRAFTOVI的使用可能会影响雄性的生育力[见非临床毒理学(13.1) ]。
8.4小儿使用
尚未在儿科患者中确定BRAFTOVI的安全性和有效性。
8.5老年人使用
在多个临床试验中,每天接受300 mg至600 mg剂量的BRAFTOVI联合比奈替尼(45 mg每天两次)的690名BRAF突变阳性黑色素瘤患者在多项临床试验中接受治疗,其中20%年龄在65-74岁之间,8%年龄在75岁以上。与年轻患者相比,老年患者未观察到BRAFTOVI加比尼替尼的安全性或有效性总体差异[见临床药理学(12.3) ]。
8.6肝功能不全
不建议在轻度肝功能不全(Child-Pugh A级)患者中调整BRAFTOVI的剂量[参见临床药理学(12.3) ]。对于中度(Child-Pugh B级)或重度(Child-Pugh C级)肝功能不全的患者,尚未确定推荐剂量。
8.7肾功能不全
对于轻至中度肾功能不全(CLcr 30至<90 mL / min)的患者,不建议调整剂量[见临床药理学(12.3) ]。对于严重肾功能不全(CLcr <30 mL / min)的患者,尚未确定推荐剂量。
10过量
由于恩可拉非尼与血浆蛋白的结合率为 86%,因此血液透析在用BRAFTOVI过量治疗中可能无效。
11说明
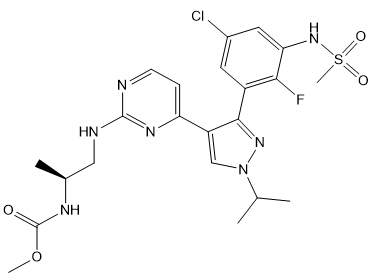
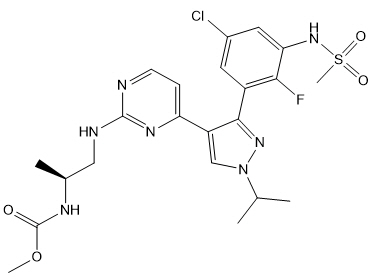
恩可拉非尼是一种激酶抑制剂。化学名称是甲基N -{(2 S)-1-[(4- {3- [5-氯-2-氟-3-(甲磺酰胺基)苯基] -1-(丙-2-基)-1 H-吡唑-4-基}嘧啶-2-基)氨基]丙烷-2-基}氨基甲酸酯。分子式为C 22 H 27 ClFN 7 O 4 S,分子量为540道尔顿。恩可拉非尼的化学结构如下所示:
恩哥拉非是白色至几乎白色的粉末。在水性介质中,encorafenib在pH 1时微溶,在pH 2时微溶,在pH 3或更高时不溶。
口服的BRAFTOVI(encorafenib)胶囊含有75 mg的encorafenib及其以下非活性成分:copovidone,poloxamer 188,微晶纤维素,琥珀酸,crospovidone,胶体二氧化硅,硬脂酸镁(植物来源)。胶囊壳包含明胶,二氧化钛,氧化铁红,氧化铁黄,四氧化三铁,会标油墨(药用釉,四氧化三铁,丙二醇)。
12临床药理学
12.1行动机制
Encorafenib是一种激酶抑制剂,在体外无细胞试验中靶向BRAF V600E以及野生型BRAF和CRAF,IC 50值分别为0.35、0.47和0.3 nM。BRAF基因中的突变(例如BRAF V600E)可导致组成型激活的BRAF激酶,从而刺激肿瘤细胞的生长。恩哥拉非尼还能够在体外与其他激酶结合,包括JNK1,JNK2,JNK3,LIMK1,LIMK2,MEK4和STK36,并在临床上可达到的浓度(≤0.9 µM)下显着降低配体与这些激酶的结合。
恩哥拉非尼抑制表达BRAF V600 E,D和K突变的肿瘤细胞系的体外生长。在植入表达BRAF V600E的肿瘤细胞的小鼠中, 恩哥拉非尼诱导了与RAF / MEK / ERK途径抑制相关的肿瘤消退。
恩哥拉非尼和比米替尼靶向RAS / RAF / MEK / ERK途径中的两个不同的激酶。与单独使用这两种药物相比,encorafenib和binimetinib的共同给药在BRAF V600E突变型人黑素瘤异种移植研究中产生了更大的体外抗BRAF突变阳性细胞系抗增殖活性,并具有更大的抗肿瘤活性。老鼠。另外,与单独使用任一药物相比,encorafenib和binimetinib 的组合延迟了小鼠BRAF V600E突变型人黑素瘤异种移植物中耐药性的出现。
12.2药效学
心脏电生理学
尚未进行评估BRAFTOVI QT延长潜力的专门研究。BRAFTOVI与剂量依赖性QTc间隔延长有关。根据一项针对成年黑色素瘤患者的QTc集中趋势分析,建议剂量的BRAFTOVI与Binimetinib联用后,与基线(ΔQTcF)相比,最大平均(90%CI)QTcF变化为18(14 22)ms [请参阅警告和注意事项(5.5) ]。
12.3药代动力学
在健康受试者和实体瘤患者中研究了encorafenib的药代动力学,这些实体瘤包括具有BRAF V600E或V600K突变的晚期和不可切除或转移性皮肤黑色素瘤。单次给药后,恩可拉非尼的全身暴露与50 mg至700 mg剂量范围内的剂量成比例。每天一次给药后,恩可拉非尼的全身暴露量小于50 mg至800 mg剂量范围内的比例剂量。在15天内达到稳态,与第1天相比,暴露量降低了50%;AUC的受试者间变异性(CV%)为12%至69%。
吸收性
口服后,恩可拉非尼的中值T max为2小时。至少86%的剂量被吸收。
食物的作用
服用100毫克BRAFTOVI单剂量(建议剂量的0.2倍)和高脂肪,高热量餐(包括大约150卡路里的蛋白质卡路里,350卡路里的碳水化合物和500卡路里的脂肪)降低了平均最大值恩可拉非尼浓度(C max)降低36%,对AUC无影响。
分配
恩考拉非在体外与人血浆蛋白结合率为 86%。血液与血浆的浓度比为0.58。表观分布体积的几何平均值(CV%)为164 L(70%)。
消除
恩哥拉非尼的平均(CV%)终末半衰期(t 1/2)为3.5小时(17%),第1天的表观清除率为14 L / h(54%),增加至32 L / h( 59%)。
代谢
主要的代谢途径是N-脱烷基化,CYP3A4是恩可拉非尼在人肝微粒体中总氧化清除的主要贡献者(83%),其次是CYP2C19(16%)和CYP2D6(1%)。
排泄
在单次口服100 mg放射性标记的恩可拉非尼后,粪便中回收了47%(不变的5%),尿液中回收了47%(不变的2%)。
特定人群
年龄(19至89岁),性别,体重,轻度肝功能不全(Child-Pugh A级)和轻度或中度肾功能不全(CLcr 30至<90 mL / min)对药代动力学没有临床意义的影响的encorafenib。尚未研究种族或种族,中度或重度肝功能不全(Child-Pugh B级或C级)和严重肾功能不全(CLcr <30 mL / min)对恩哥拉非尼药代动力学的影响。
药物相互作用研究
临床研究
CYP3A4抑制剂对Encorafenib:强烈(泊沙康唑)合用或中度(地尔硫卓)CYP3A4抑制剂BRAFTOVI增加的AUC encorafenib由3-和2倍,分别,而且增加了Ç 最大由68%和45%,分别服用一次BRAFTOVI 50 mg(建议剂量的0.1倍)后。
CYP3A4诱导剂对Encorafenib的影响:尚未研究CYP3A4诱导剂与encorafenib的并用影响。在临床试验中,稳定状态的encorafenib暴露量低于encorafenib首次给药后的暴露量,提示CYP3A4自动诱导。
减酸剂对恩可拉非尼的影响:并用质子泵抑制剂雷贝拉唑对恩可拉非尼的 AUC和C max无影响。
联合治疗: BRAFTOVI(UGT1A1抑制剂)与比美替尼(UGT1A1底物)的共同给药对比美替尼的暴露无影响。
体外研究
的效果Encorafenib对CYP / UGT衬底:Encorafenib是UGT1A1,CYP1A2,CYP2B6,CYP2C8 / 9,CYP2D6,和CYP3A的可逆抑制剂,和CYP3A4的临床相关的血浆浓度的时间依赖性抑制剂。在临床相关的血浆浓度下,Encorafenib诱导CYP2B6,CYP2C9和CYP3A4。
转运蛋白对Encorafenib的影响:Encorafenib是P-糖蛋白(P-gp)的底物。在临床相关血浆浓度下,Encorafenib不是乳腺癌抗性蛋白(BCRP),多药抗性相关蛋白2(MRP2),有机阴离子转运多肽(OATP1B1,OATP1B3)或有机阳离子转运蛋白(OCT1)的底物。
的效果Encorafenib上转运:Encorafenib抑制的P-gp,BCRP,OCT2,有机阴离子转运体(OAT1,OAT3),OATP1B1,和OATP1B3,但不OCT1或MRP2在临床相关的血浆浓度。
13毒理学
13.1致癌,诱变,生育力受损
尚未进行encorafenib的致癌性研究。在评估细菌的反向突变,哺乳动物细胞中的染色体畸变或大鼠骨髓中的微核的研究中,Encorafenib没有遗传毒性。
恩考拉非未在动物中进行专门的生育力研究。在一项针对大鼠的一般毒理学研究中,以450毫克临床剂量(基于AUC)的剂量,其暴露量约为人体暴露量的13倍,从而降低了睾丸和附睾的重量,睾丸的肾小管变性以及附睾中的少精症。在任何非人类的灵长类动物毒性研究中,未发现对任何性别的生殖器官都有影响。
13.2动物毒理学和/或药理学
在Encorafenib剂量为20 mg / kg /天(在基于AUC的450 mg临床剂量下,人体暴露量的14倍左右)或更高剂量的情况下,在4周和13周内,大鼠的胃部发生了增生和角化过度的不良组织病理学发现学习。
14临床研究
在一项随机,主动控制,开放标签,多中心试验(COLUMBUS; NCT01909453)中评估了BRAFTOVI与Binimetinib的组合。使用bioMerieux THxID™BRAF分析检测出的合格患者必须患有BRAF V600E或V600K突变阳性的不可切除或转移性黑色素瘤。允许患者在辅助条件下接受免疫治疗,并在先前的免疫治疗方案中接受不可切除的局部晚期或转移性疾病的治疗。禁止事先使用BRAF抑制剂或MEK抑制剂。根据美国癌症联合委员会(AJCC)阶段(IIIB,IIIC,IVM1a或IVM1b,对比IVM1c),东部合作肿瘤小组(ECOG)的表现状态(0对1)以及先前针对无法切除或转移性疾病的免疫疗法(是与否)。
患者被随机分配(1:1:1:1),以接受每日一次BRAFTOVI 450 mg联合比米替尼45 mg每天两次(BRAFTOVI与Binimetinib联合),BRAFTOVI 300 mg每天一次或维拉非尼960 mg每天两次。继续治疗直至疾病进展或不可接受的毒性。下面仅描述批准的给药结果(BRAFTOVI 450 mg与比美替尼45 mg组合)。
主要的疗效结局指标是无进展生存期(PFS),通过盲法独立中央评价进行评估,以比较BRAFTOVI与Binimetinib与vemurafenib的组合。其他功效结局指标包括总体生存率(OS)以及客观缓解率(ORR)和缓解持续时间(DoR),这些均通过集中评估进行评估。
总共577例患者被随机分配,其中192例联合比尼替尼组与BRAFTOVI联合使用,194例联合BRAFTOVI臂与191联合vemurafenib联合使用。在383例随机分组接受BRAFTOVI联合比尼替尼或维拉非尼治疗的患者中,中位年龄为56岁(20至89岁),其中男性为59%,白人为91%,基线ECOG表现为72%。 0. 95%(95%)有转移性疾病,65%为IVM1c期,4%接受了先前的CTLA-4,PD-1或PD-L1定向抗体。28%(28%)的基线血清乳酸脱氢酶(LDH)升高,45%的基线≥3个器官受累于肿瘤,3%的患者发生脑转移。根据集中测试,100%的患者肿瘤BRAF突变检测为阳性;BRAF V600E(88%),BRAF V600K(11%)或两者(<1%)。
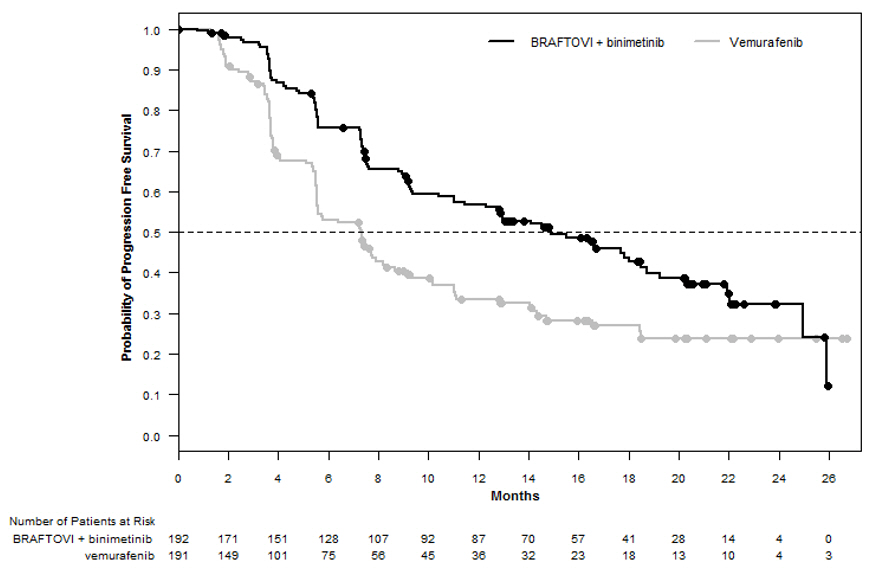
与维莫非尼相比,BRAFTOVI与比尼替尼联合使用在PFS上具有统计学上的显着改善。疗效结果总结在表6和图1中。
表6:COLUMBUS的疗效结果

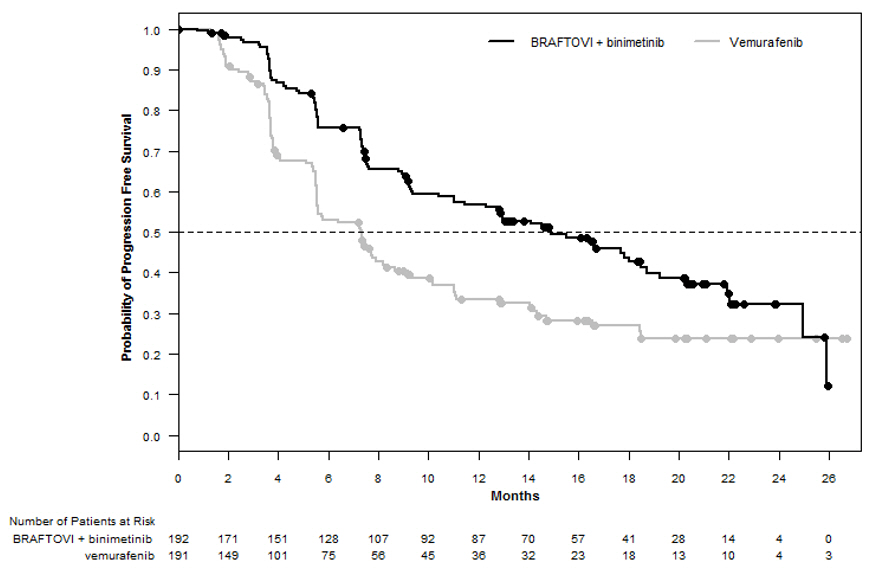
图1:哥伦布无进展生存期的Kaplan-Meier曲线
16供应/存储和处理方式
BRAFTOVI(encorafenib)以75 mg硬明胶胶囊的形式提供。
75 mg:米色瓶盖上的程式化“ A”,白色机身上的“ LGX 75mg”,以纸箱(NDC 70255-025-01)的形式提供,每箱包含两瓶90粒胶囊(NDC 70255-025-02)和纸箱(NDC 70255) -025-03),每瓶包含两瓶60粒胶囊(NDC 70255-025-04)。
存放在20°C至25°C(68°F至77°F); 允许在15°C至30°C(59°F至86°F)之间的偏移[请参阅USP控制的室温]。如果盖下的安全密封件损坏或缺失,请不要使用。分配原瓶。不要去除干燥剂。防潮。保持容器密闭。
17患者咨询信息
建议患者阅读FDA批准的患者标签(用药指南)。
通知患者以下内容:
新的原发性皮肤恶性肿瘤
劝告患者立即联系其医疗保健提供者,以发现新的皮肤病变的变化或发展[见警告和注意事项(5.1) ]。
出血
劝告患者任何暗示出血的症状(例如异常出血)立即通知其医疗人员[请参阅警告和注意事项(5.3) ]。
葡萄膜炎
如果患者视力有任何变化,建议他们联系其医疗保健提供者[请参阅警告和注意事项(5.4) ]。
QT延长
告知患者BRAFTOVI会导致QTc间隔延长症状,并告知医生是否有任何QTc间隔延长症状,例如晕厥[ 见警告和注意事项(5.5) ]。
生殖潜力的男性和女性
胚胎-胎儿毒性:建议具有生殖潜能的女性对胎儿有潜在危险。建议有生育潜力的女性在用BRAFTOVI治疗期间和最终剂量后2周内使用有效的非激素避孕方法。建议女性在接受BRAFTOVI治疗期间怀孕或怀疑怀孕时联系其医疗保健提供者[见警告和注意事项(5.6),在特定人群中使用(8.1) ]。
哺乳期:建议妇女在用BRAFTOVI治疗期间和最终剂量后2周内不要母乳喂养[见在特定人群中使用(8.2) ]。
不育:建议有生殖能力的男性认为BRAFTOVI可能会损害生育能力[见在特定人群中的使用(8.3) ]。
强或中度CYP3A诱导剂或抑制剂
将BRAFTOVI与强或中度CYP3A抑制剂并用可能会增加encorafenib的浓度; 而BRAFTOVI与强或中度CYP3A诱导剂的共同给药可能会降低encorafenib的浓度。告知患者在服用BRAFTOVI时需要避免使用某些药物,并告知其医疗保健提供者所有伴随药物,包括处方药,非处方药,维生素和草药产品。建议患者在服用BRAFTOVI时避免吃葡萄柚或葡萄柚汁[见药物相互作用(7.1) ]。
存储
BRAFTOVI对水分敏感。建议患者将BRAFTOVI存放在带有干燥剂的原始瓶中,并保持瓶盖紧密关闭。请勿从瓶中取出干燥剂。
分配者:
Array BioPharma Inc.
核桃木街3200号
,科罗拉多州80301©2018 Array BioPharma Inc.保留所有权利。
BRAFTOVI ®是阵列生物制药公司在美国及其他国家的注册商标。
获得专利。参见www.arraybiopharma.com/patents



主要显示面板-75毫克胶囊瓶纸箱-025-01
NDC 70255-025-01
BRAFTOVI ®
(encorafenib)胶囊75毫克
仅限Rx
2瓶x 90粒
主要显示面板-75毫克胶囊瓶纸箱-025-03
NDC 70255-025-03
BRAFTOVI ®
(encorafenib)胶囊75毫克
仅限Rx
2瓶x 60胶囊
主要显示面板-50毫克胶囊瓶纸箱
NDC 70255-020-01
BRAFTOVI™
(encorafenib)胶囊
50毫克胶囊仅限Rx
2瓶x 60胶囊
【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
本说明书来源于:美国FDA
https://nctr-crs.fda.gov/fdalabel/services/spl/set-ids/235dfc38-0f0b-4037-b501-7a9f4294740c/spl-doc?hl=encorafenib
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书。
1 INDICATIONS AND USAGE
BRAFTOVI® is indicated, in combination with binimetinib, for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test [see Dosage and Administration (2.1)].
Limitations of Use
BRAFTOVI is not indicated for treatment of patients with wild-type BRAF melanoma [see Warnings and Precautions (5.2)].
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Confirm the presence of a BRAF V600E or V600K mutation in tumor specimens prior to initiating BRAFTOVI [see Warnings and Precautions (5.2), Clinical Studies (14)]. Information on FDA-approved tests for the detection of BRAF V600E and V600K mutations in melanoma is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of BRAFTOVI is 450 mg (six 75 mg capsules) orally taken once daily in combination with binimetinib until disease progression or unacceptable toxicity. Refer to the binimetinib prescribing information for recommended binimetinib dosing information.
BRAFTOVI may be taken with or without food [see Clinical Pharmacology (12.3)]. Do not take a missed dose of BRAFTOVI within 12 hours of the next dose of BRAFTOVI.
Do not take an additional dose if vomiting occurs after BRAFTOVI administration but continue with the next scheduled dose.
2.3 Dosage Modifications for Adverse Reactions
If binimetinib is withheld, reduce BRAFTOVI to a maximum dose of 300 mg once daily until binimetinib is resumed [see Warnings and Precautions (5.7)].
Dose reductions for adverse reactions associated with BRAFTOVI are presented in Table 1.
Table 1: Recommended Dose Reductions for BRAFTOVI for Adverse Reactions

Dosage modifications for adverse reactions associated with BRAFTOVI are presented in Table 2.
Table 2: Recommended Dosage Modifications for BRAFTOVI for Adverse Reactions
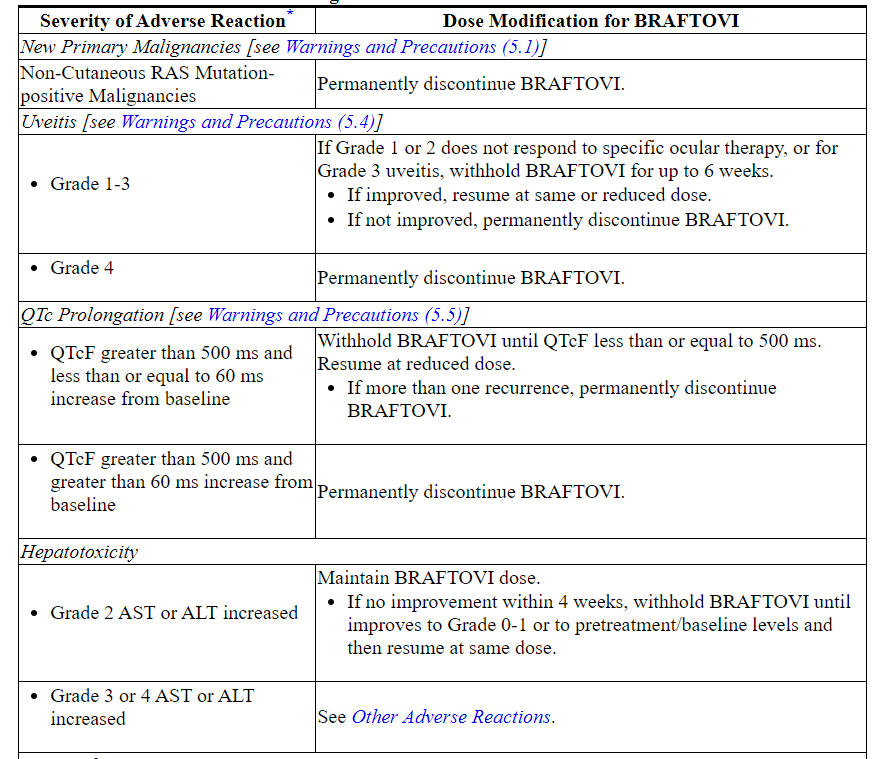
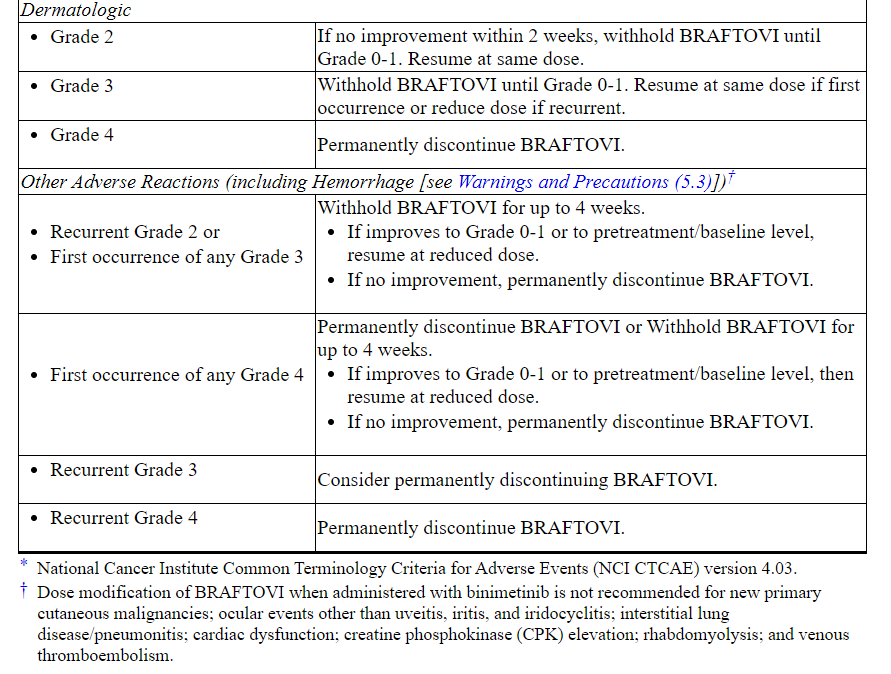
Refer to the binimetinib prescribing information for dose modifications for adverse reactions associated with binimetinib.
2.4 Dose Modifications for Coadministration with Strong or Moderate CYP3A4 Inhibitors
Avoid coadministration with strong or moderate CYP3A4 inhibitors during treatment with BRAFTOVI. If coadministration with a strong or moderate CYP3A4 inhibitor is unavoidable, reduce the BRAFTOVI dose according to the recommendations in Table 3. After the inhibitor has been discontinued for 3 to 5 elimination half-lives, resume the BRAFTOVI dose that was taken prior to initiating the CYP3A4 inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Table 3: Recommended Dose Reductions for BRAFTOVI for Coadministration with Strong or Moderate CYP3A4 Inhibitors
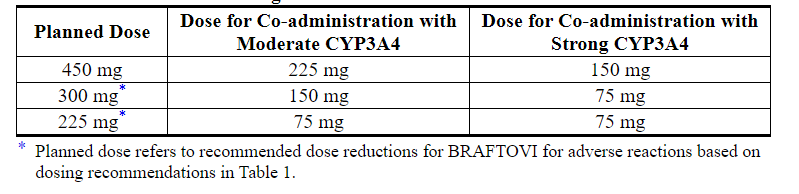
3 DOSAGE FORMS AND STRENGTHS
Capsules: 75 mg, hard gelatin, stylized "A" on beige cap and "LGX 75mg" on white body
4 CONTRAINDICATIONS
None.
5 WARNINGS AND PRECAUTIONS
5.1 New Primary Malignancies
New primary malignancies, cutaneous and non-cutaneous, have been observed in patients treated with BRAF inhibitors and can occur with BRAFTOVI.
Cutaneous Malignancies
In COLUMBUS, cutaneous squamous cell carcinoma (cuSCC), including keratoacanthoma (KA), occurred in 2.6%, and basal cell carcinoma occurred in 1.6% of patients who received BRAFTOVI in combination with binimetinib. Median time to first occurrence of cuSCC/KA was 5.8 months (range 1 to 9 months) [see Adverse Reactions (6.1)].
For patients who received BRAFTOVI as a single agent, cuSCC/KA was reported in 8%, basal cell carcinoma in 1%, and a new primary melanoma in 5% of patients.
Perform dermatologic evaluations prior to initiating treatment, every 2 months during treatment, and for up to 6 months following discontinuation of treatment. Manage suspicious skin lesions with excision and dermatopathologic evaluation. Dose modification is not recommended for new primary cutaneous malignancies.
Non-Cutaneous Malignancies
Based on its mechanism of action, BRAFTOVI may promote malignancies associated with activation of RAS through mutation or other mechanisms [see Warnings and Precautions (5.2)]. Monitor patients receiving BRAFTOVI for signs and symptoms of non-cutaneous malignancies. Discontinue BRAFTOVI for RAS mutation-positive non-cutaneous malignancies [see Dosage and Administration (2.3)].
5.2 Tumor Promotion in BRAF Wild-Type Tumors
In vitro experiments have demonstrated paradoxical activation of MAP-kinase signaling and increased cell proliferation in BRAF wild-type cells, which are exposed to BRAF inhibitors. Confirm evidence of BRAF V600E or V600K mutation prior to initiating BRAFTOVI [see Indications and Usage (1), Dosage and Administration (2.1)].
5.3 Hemorrhage
Hemorrhage can occur when BRAFTOVI is administered in combination with binimetinib. In COLUMBUS, hemorrhage occurred in 19% of patients receiving BRAFTOVI in combination with binimetinib; Grade 3 or greater hemorrhage occurred in 3.2% of patients. The most frequent hemorrhagic events were gastrointestinal, including rectal hemorrhage (4.2%), hematochezia (3.1%), and hemorrhoidal hemorrhage (1%). Fatal intracranial hemorrhage in the setting of new or progressive brain metastases occurred in 1.6% of patients.
Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.4 Uveitis
Uveitis, including iritis and iridocyclitis, has been reported in patients treated with BRAFTOVI in combination with binimetinib. In COLUMBUS, the incidence of uveitis among patients treated with BRAFTOVI in combination with binimetinib was 4%.
Assess for visual symptoms at each visit. Perform an ophthalmologic evaluation at regular intervals and for new or worsening visual disturbances, and to follow new or persistent ophthalmologic findings. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.5 QT Prolongation
BRAFTOVI is associated with dose-dependent QTc interval prolongation in some patients [see Clinical Pharmacology (12.2)]. In COLUMBUS, an increase in QTcF to > 500 ms was measured in 0.5% (1/192) of patients who received BRAFTOVI in combination with binimetinib.
Monitor patients who already have or who are at significant risk of developing QTc prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, severe or uncontrolled heart failure and those taking other medicinal products associated with QT prolongation. Correct hypokalemia and hypomagnesemia prior to and during BRAFTOVI administration. Withhold, reduce dose, or permanently discontinue for QTc > 500 ms [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.6 Embryo-Fetal Toxicity
Based on its mechanism of action, BRAFTOVI can cause fetal harm when administered to a pregnant woman. Encorafenib produced embryo-fetal developmental changes in rats and rabbits and was an abortifacient in rabbits at doses greater than or equal to those resulting in exposures approximately 26 (in the rat) and 178 (in the rabbit) times the human exposure at the recommended dose of 450 mg, with no clear findings at lower doses.
Advise women of the potential risk to a fetus. Advise females of reproductive potential to use an effective, non-hormonal method of contraception since BRAFTOVI can render hormonal contraceptives ineffective, during treatment and for 2 weeks after the final dose of BRAFTOVI [see Use in Specific Populations (8.1, 8.3)].
5.7 Risks Associated with BRAFTOVI as a Single Agent
BRAFTOVI when used as a single agent is associated with an increased risk of certain adverse reactions compared to when BRAFTOVI is used in combination with binimetinib. Grades 3 or 4 dermatologic reactions occurred in 21% of patients treated with BRAFTOVI single agent compared to 2% of patients treated with BRAFTOVI in combination with binimetinib [see Warnings and Precautions (5.1), Adverse Reactions (6.1)].
If binimetinib is temporarily interrupted or permanently discontinued, reduce the dose of BRAFTOVI as recommended [see Dosage and Administration (2.3)].
5.8 Risks Associated with Combination Treatment
BRAFTOVI is indicated for use in combination with binimetinib. Refer to the binimetinib prescribing information for additional risk information that applies to combination use treatment.
6 ADVERSE REACTIONS
The following adverse reactions are described elsewhere in the labeling:
- New Primary Malignancies [see Warnings and Precautions (5.1)]
- Hemorrhage [see Warnings and Precautions (5.3)]
- Uveitis [see Warnings and Precautions (5.4)]
- QT Prolongation [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of BRAFTOVI in combination with binimetinib is described in 192 patients with BRAF V600 mutation-positive unresectable or metastatic melanoma who received BRAFTOVI (450 mg once daily) in combination with binimetinib (45 mg twice daily) in a randomized open-label, active-controlled trial (COLUMBUS).
The COLUMBUS trial [see Clinical Studies (14)] excluded patients with a history of Gilbert's syndrome, abnormal left ventricular ejection fraction, prolonged QTc (>480 msec), uncontrolled hypertension, and history or current evidence of retinal vein occlusion. The median duration of exposure was 11.8 months for patients treated with BRAFTOVI in combination with binimetinib and 6.2 months for patients treated with vemurafenib.
The most common (≥ 25%) adverse reactions in patients receiving BRAFTOVI in combination with binimetinib were fatigue, nausea, vomiting, abdominal pain, and arthralgia.
Adverse reactions leading to dose interruptions of BRAFTOVI occurred in 30% of patients receiving BRAFTOVI in combination with binimetinib; the most common were nausea (7%), vomiting (7%) and pyrexia (4%). Adverse reactions leading to dose reductions of BRAFTOVI occurred in 14% of patients receiving BRAFTOVI in combination with binimetinib; the most common were arthralgia (2%), fatigue (2%) and nausea (2%). Five percent (5%) of patients receiving BRAFTOVI in combination with binimetinib experienced an adverse reaction that resulted in permanent discontinuation of BRAFTOVI; the most common were hemorrhage in 2% and headache in 1% of patients.
Table 4 and Table 5 present adverse drug reactions and laboratory abnormalities, respectively, identified in COLUMBUS. The COLUMBUS trial was not designed to demonstrate a statistically significant difference in adverse reaction rates for BRAFTOVI in combination with binimetinib, as compared to vemurafenib, for any specific adverse reaction listed in Table 4.
Table 4: Adverse Reactions Occurring in ≥ 10% of Patients Receiving BRAFTOVI in Combination with Binimetinib in COLUMBUS*
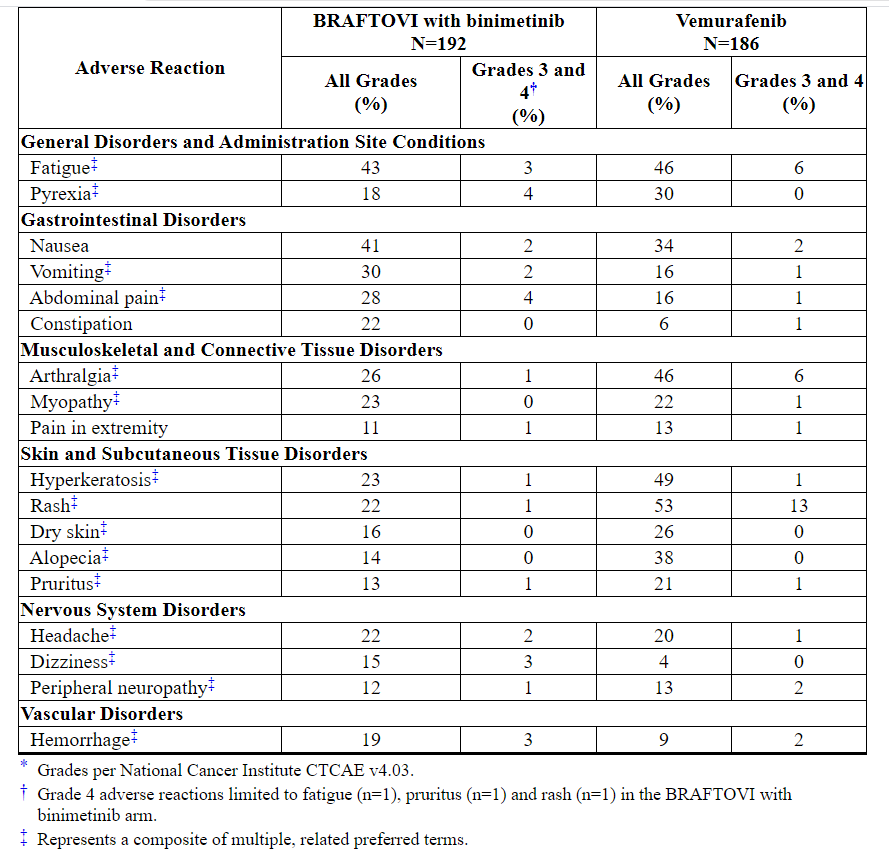
BRAFTOVI when used as a single agent increases the risk of certain adverse reactions compared to BRAFTOVI in combination with binimetinib. In patients receiving BRAFTOVI 300 mg orally once daily as a single agent, the following adverse reactions were observed at a higher rate (≥ 5%) compared to patients receiving BRAFTOVI in combination with binimetinib: palmar-plantar erythrodysesthesia syndrome (51% vs. 7%), hyperkeratosis (57% vs. 23%), dry skin (38% vs. 16%), erythema (16% vs. 7%), rash (41% vs. 22%), alopecia (56% vs. 14%), pruritus (31% vs. 13%), arthralgia (44% vs. 26%), myopathy (33% vs. 23%), back pain (15% vs. 9%), dysgeusia (13% vs. 6%), and acneiform dermatitis (8% vs. 3%).
Other clinically important adverse reactions occurring in < 10% of patients who received BRAFTOVI in combination with binimetinib were:
Nervous system disorders: Facial paresis
Gastrointestinal disorders: Pancreatitis
Skin and subcutaneous tissue disorders: Panniculitis
Immune system disorders: Drug hypersensitivity
Table 5: Laboratory Abnormalities Occurring in ≥ 10% (All Grades) of Patients Receiving BRAFTOVI in Combination with Binimetinib in COLUMBUS*
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on BRAFTOVI
Strong or Moderate CYP3A4 Inhibitors
Concomitant administration of BRAFTOVI with a strong or moderate CYP3A4 inhibitor increased encorafenib plasma concentrations and may increase encorafenib adverse reactions [see Clinical Pharmacology (12.3)]. Avoid coadministration of BRAFTOVI with strong or moderate CYP3A4 inhibitors, including grapefruit juice. If coadministration of strong or moderate CYP3A4 inhibitors cannot be avoided, modify dose as recommended [see Dosage and Administration (2.4)].
Strong or Moderate CYP3A4 Inducers
Concomitant administration of BRAFTOVI with a strong or moderate CYP3A4 inducer may decrease encorafenib plasma concentrations and may decrease encorafenib efficacy [see Clinical Pharmacology (12.3)]. Avoid concomitant administration of strong or moderate CYP3A4 inducers with BRAFTOVI.
7.2 Effect of BRAFTOVI on Other Drugs
Sensitive CYP3A4 Substrates
Concomitant administration of BRAFTOVI with sensitive CYP3A4 substrates may result in increased toxicity or decreased efficacy of these agents.
Coadministration of BRAFTOVI with hormonal contraceptives (CYP3A4 substrates) can result in decreased concentrations and loss of hormonal contraceptive efficacy. Avoid hormonal contraceptives [see Use in Specific Populations (8.3)].
7.3 Drugs That Prolong the QT Interval
BRAFTOVI is associated with dose-dependent QTc interval prolongation. Avoid coadministration of BRAFTOVI with medicinal products with a known potential to prolong QT/QTc interval [see Warnings and Precautions (5.5), Clinical Pharmacology (12.2)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, BRAFTOVI can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available clinical data on the use of BRAFTOVI during pregnancy. In animal reproduction studies, encorafenib produced embryo-fetal developmental changes in rats and rabbits and was an abortifacient in rabbits at doses greater than or equal to those resulting in exposures approximately 26 (in the rat) and 178 (in the rabbit) times the human exposure at the clinical dose of 450 mg, with no clear findings at lower doses (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In reproductive toxicity studies, administration of encorafenib to rats during the period of organogenesis resulted in maternal toxicity, decreased fetal weights, and increased incidence of total skeletal variations at a dose of 20 mg/kg/day (approximately 26 times the human exposure based on area under the concentration-time curve [AUC] at the recommended clinical dose of 450 mg once daily). In pregnant rabbits, administration of encorafenib during the period of organogenesis resulted in maternal toxicity, decreased fetal body weights, increased incidence of total skeletal variations and increased post-implantation loss, including total loss of pregnancy at a dose of 75 mg/kg/day (approximately 178 times the human exposure based on AUC at the recommended clinical dose of 450 mg once daily). While formal placental transfer studies have not been performed, encorafenib exposure in the fetal plasma of both rats and rabbits was up to 1.7% and 0.8%, respectively, of maternal exposure.
8.2 Lactation
Risk Summary
There are no data on the presence of encorafenib or its metabolites in human milk or the effects of encorafenib on the breastfed infant, or on milk production. Because of the potential for serious adverse reactions from BRAFTOVI in breastfed infants, advise women not to breastfeed during treatment with BRAFTOVI and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating BRAFTOVI [see Use in Specific Populations (8.1)].
Contraception
BRAFTOVI can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise females of reproductive potential to use effective contraception during treatment with BRAFTOVI and for 2 weeks after the final dose. Counsel patients to use a non-hormonal method of contraception since BRAFTOVI has the potential to render hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Infertility
Males
Based on findings in male rats at doses approximately 13 times the human exposure at the 450 mg clinical dose, use of BRAFTOVI may impact fertility in males [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of BRAFTOVI have not been established in pediatric patients.
8.5 Geriatric Use
Of the 690 patients with BRAF mutation-positive melanoma who received BRAFTOVI at doses between 300 mg and 600 mg once daily in combination with binimetinib (45 mg twice daily) across multiple clinical trials, 20% were aged 65 to 74 years and 8% were aged 75 years and older. No overall differences in the safety or effectiveness of BRAFTOVI plus binimetinib were observed in elderly patients as compared to younger patients [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
Dose adjustment for BRAFTOVI is not recommended in patients with mild hepatic impairment (Child-Pugh Class A) [see Clinical Pharmacology (12.3)]. A recommended dose has not been established for patients with moderate (Child-Pugh Class B) or severe (Child-Pugh Class C) hepatic impairment.
8.7 Renal Impairment
No dose adjustment is recommended for patients with mild to moderate renal impairment (CLcr 30 to < 90 mL/min) [see Clinical Pharmacology (12.3)]. A recommended dose has not been established for patients with severe renal impairment (CLcr < 30 mL/min).
10 OVERDOSAGE
Since encorafenib is 86% bound to plasma proteins, hemodialysis is likely to be ineffective in the treatment of overdose with BRAFTOVI.
11 DESCRIPTION
Encorafenib is a kinase inhibitor. The chemical name is methyl N-{(2S)-1-[(4-{3-[5-chloro-2-fluoro-3-(methanesulfonamido)phenyl]-1-(propan-2-yl)-1H-pyrazol-4-yl}pyrimidin-2-yl)amino]propan-2-yl}carbamate. The molecular formula is C22H27ClFN7O4S and the molecular weight is 540 daltons. The chemical structure of encorafenib is shown below:
Encorafenib is a white to almost white powder. In aqueous media, encorafenib is slightly soluble at pH 1, very slightly soluble at pH 2, and insoluble at pH 3 and higher.
BRAFTOVI (encorafenib) capsules for oral use contain 75 mg of encorafenib with the following inactive ingredients: copovidone, poloxamer 188, microcrystalline cellulose, succinic acid, crospovidone, colloidal silicon dioxide, magnesium stearate (vegetable origin). The capsule shell contains gelatin, titanium dioxide, iron oxide red, iron oxide yellow, ferrosoferric oxide, monogramming ink (pharmaceutical glaze, ferrosoferric oxide, propylene glycol).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Encorafenib is a kinase inhibitor that targets BRAF V600E, as well as wild-type BRAF and CRAF in in vitro cell-free assays with IC50 values of 0.35, 0.47, and 0.3 nM, respectively. Mutations in the BRAF gene, such as BRAF V600E, can result in constitutively activated BRAF kinases that may stimulate tumor cell growth. Encorafenib was also able to bind to other kinases in vitro including JNK1, JNK2, JNK3, LIMK1, LIMK2, MEK4, and STK36 and substantially reduce ligand binding to these kinases at clinically achievable concentrations (≤ 0.9 µM).
Encorafenib inhibited in vitro growth of tumor cell lines expressing BRAF V600 E, D, and K mutations. In mice implanted with tumor cells expressing BRAF V600E, encorafenib induced tumor regressions associated with RAF/MEK/ERK pathway suppression.
Encorafenib and binimetinib target two different kinases in the RAS/RAF/MEK/ERK pathway. Compared with either drug alone, co-administration of encorafenib and binimetinib resulted in greater anti-proliferative activity in vitro in BRAF mutation-positive cell lines and greater anti-tumor activity with respect to tumor growth inhibition in BRAF V600E mutant human melanoma xenograft studies in mice. Additionally, the combination of encorafenib and binimetinib delayed the emergence of resistance in BRAF V600E mutant human melanoma xenografts in mice compared to either drug alone.
12.2 Pharmacodynamics
Cardiac Electrophysiology
A dedicated study to evaluate the QT prolongation potential of BRAFTOVI has not been conducted. BRAFTOVI is associated with dose-dependent QTc interval prolongation. Following administration of the recommended dose of BRAFTOVI in combination with binimetinib, based on a central tendency analysis of QTc in a study of adult patients with melanoma, the largest mean (90% CI) QTcF change from baseline (ΔQTcF) was 18 (14 to 22) ms [see Warnings and Precautions (5.5)].
12.3 Pharmacokinetics
The pharmacokinetics of encorafenib were studied in healthy subjects and patients with solid tumors, including advanced and unresectable or metastatic cutaneous melanoma harboring a BRAF V600E or V600K mutation. After a single dose, systemic exposure of encorafenib was dose proportional over the dose range of 50 mg to 700 mg. After once-daily dosing, systemic exposure of encorafenib was less than dose proportional over the dose range of 50 mg to 800 mg. Steady-state was reached within 15 days, with exposure being 50% lower compared to Day 1; intersubject variability (CV%) of AUC ranged from 12% to 69%.
Absorption
After oral administration, the median Tmax of encorafenib is 2 hours. At least 86% of the dose is absorbed.
Effect of Food
Administration of a single dose of BRAFTOVI 100 mg (0.2 times the recommended dose) with a high-fat, high-calorie meal (comprised of approximately 150 calories from protein, 350 calories from carbohydrates, and 500 calories from fat) decreased the mean maximum encorafenib concentration (Cmax) by 36% with no effect on AUC.
Distribution
Encorafenib is 86% bound to human plasma proteins in vitro. The blood-to-plasma concentration ratio is 0.58. The geometric mean (CV%) of apparent volume of distribution is 164 L (70%).
Elimination
The mean (CV%) terminal half-life (t1/2) of encorafenib is 3.5 hours (17%), and the apparent clearance is 14 L/h (54%) at day 1, increasing to 32 L/h (59%) at steady-state.
Metabolism
The primary metabolic pathway is N-dealkylation, with CYP3A4 as the main contributor (83%) to total oxidative clearance of encorafenib in human liver microsomes, followed by CYP2C19 (16%) and CYP2D6 (1%).
Excretion
Following a single oral dose of 100 mg radiolabeled encorafenib, 47% (5% unchanged) of the administered dose was recovered in the feces and 47% (2% unchanged) was recovered in the urine.
Specific Populations
Age (19 to 89 years), sex, body weight, mild hepatic impairment (Child-Pugh Class A), and mild or moderate renal impairment (CLcr 30 to < 90 mL/min) do not have a clinically meaningful effect on the pharmacokinetics of encorafenib. The effect of race or ethnicity, moderate or severe hepatic impairment (Child-Pugh Class B or C), and severe renal impairment (CLcr < 30 mL/min) on encorafenib pharmacokinetics have not been studied.
Drug Interaction Studies
Clinical Studies
Effect of CYP3A4 Inhibitors on Encorafenib: Coadministration of a strong (posaconazole) or moderate (diltiazem) CYP3A4 inhibitor with BRAFTOVI increased the AUC of encorafenib by 3- and 2-fold, respectively, and increased the Cmax by 68% and 45%, respectively, after a single BRAFTOVI dose of 50 mg (0.1 times the recommended dose).
Effect of CYP3A4 Inducers on Encorafenib: The effect of coadministration of a CYP3A4 inducer on encorafenib exposure has not been studied. In clinical trials, steady-state encorafenib exposures were lower than encorafenib exposures after the first dose, suggesting CYP3A4 auto-induction.
Effect of Acid Reducing Agents on Encorafenib: Coadministration of a proton pump inhibitor, rabeprazole, had no effect on AUC and Cmax of encorafenib.
Combination Treatment: Coadministration of BRAFTOVI (UGT1A1 inhibitor) with binimetinib (UGT1A1 substrate) had no effect on binimetinib exposure.
In Vitro Studies
Effect of Encorafenib on CYP/UGT Substrates: Encorafenib is a reversible inhibitor of UGT1A1, CYP1A2, CYP2B6, CYP2C8/9, CYP2D6, and CYP3A, and a time-dependent inhibitor of CYP3A4 at clinically relevant plasma concentrations. Encorafenib induced CYP2B6, CYP2C9, and CYP3A4 at clinically relevant plasma concentrations.
Effect of Transporters on Encorafenib: Encorafenib is a substrate of P-glycoprotein (P-gp). Encorafenib is not a substrate of breast cancer resistance protein (BCRP), multidrug resistance-associated protein 2 (MRP2), organic anion transporting polypeptide (OATP1B1, OATP1B3) or organic cation transporter (OCT1) at clinically relevant plasma concentrations.
Effect of Encorafenib on Transporters: Encorafenib inhibited P-gp, BCRP, OCT2, organic anion transporter (OAT1, OAT3), OATP1B1, and OATP1B3, but not OCT1 or MRP2 at clinically relevant plasma concentrations.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with encorafenib have not been conducted. Encorafenib was not genotoxic in studies evaluating reverse mutations in bacteria, chromosomal aberrations in mammalian cells, or micronuclei in bone marrow of rats.
No dedicated fertility studies were performed with encorafenib in animals. In a general toxicology study in rats, decreased testes and epididymis weights, tubular degeneration in testes, and oligospermia in epididymides were observed at doses approximately 13 times the human exposure at the 450 mg clinical dose based on AUC. No effects on reproductive organs were observed in either sex in any of the non-human primate toxicity studies.
13.2 Animal Toxicology and/or Pharmacology
Adverse histopathology findings of hyperplasia and hyperkeratosis occurred in the stomach of rats at encorafenib doses of 20 mg/kg/day (approximately 14 times the human exposure at the 450 mg clinical dose based on AUC) or greater, in both 4 and 13-week studies.
14 CLINICAL STUDIES
BRAFTOVI in combination with binimetinib was evaluated in a randomized, active-controlled, open-label, multicenter trial (COLUMBUS; NCT01909453). Eligible patients were required to have BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma, as detected using the bioMerieux THxID™BRAF assay. Patients were permitted to have received immunotherapy in the adjuvant setting and one prior line of immunotherapy for unresectable locally advanced or metastatic disease. Prior use of BRAF inhibitors or MEK inhibitors was prohibited. Randomization was stratified by American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, versus IVM1c), Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1), and prior immunotherapy for unresectable or metastatic disease (yes versus no).
Patients were randomized (1:1:1) to receive BRAFTOVI 450 mg once daily in combination with binimetinib 45 mg twice daily (BRAFTOVI in combination with binimetinib), BRAFTOVI 300 mg once daily, or vemurafenib 960 mg twice daily. Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved dosing (BRAFTOVI 450 mg in combination with binimetinib 45 mg) are described below.
The major efficacy outcome measure was progression-free survival (PFS), as assessed by a blinded independent central review, to compare BRAFTOVI in combination with binimetinib with vemurafenib. Additional efficacy outcome measures included overall survival (OS), as well as objective response rate (ORR) and duration of response (DoR) which were assessed by central review.
A total of 577 patients were randomized, 192 to the BRAFTOVI in combination with binimetinib arm, 194 to the BRAFTOVI arm, and 191 to the vemurafenib arm. Of the 383 patients randomized to either the BRAFTOVI in combination with binimetinib or the vemurafenib arms, the median age was 56 years (20 to 89 years), 59% were male, 91% were White, and 72% had baseline ECOG performance status of 0. Ninety-five percent (95%) had metastatic disease, 65% were Stage IVM1c, and 4% received prior CTLA-4, PD-1, or PD-L1 directed antibodies. Twenty-eight percent (28%) had elevated baseline serum lactate dehydrogenase (LDH), 45% had ≥ 3 organs with tumor involvement at baseline, and 3% had brain metastases. Based on centralized testing, 100% of patients' tumors tested positive for BRAF mutations; BRAF V600E (88%), BRAF V600K (11%), or both (<1%).
BRAFTOVI in combination with binimetinib demonstrated a statistically significant improvement in PFS compared to vemurafenib. Efficacy results are summarized in Table 6 and Figure 1.
Table 6: Efficacy Results for COLUMBUS
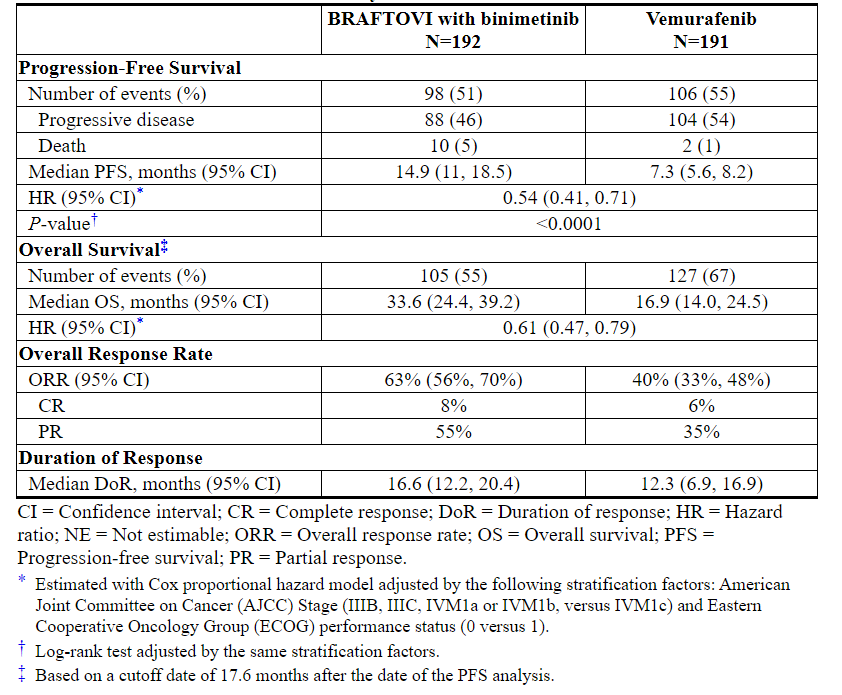
Figure 1: Kaplan-Meier Curves for Progression-Free Survival in COLUMBUS
16 HOW SUPPLIED/STORAGE AND HANDLING
BRAFTOVI (encorafenib) is supplied as 75 mg hard gelatin capsules.
75 mg: stylized "A" on beige cap and "LGX 75mg" on white body, available in cartons (NDC 70255-025-01) containing two bottles of 90 capsules each (NDC 70255-025-02) and cartons (NDC 70255-025-03) containing two bottles of 60 capsules each (NDC 70255-025-04).
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. Do not use if safety seal under cap is broken or missing. Dispense in original bottle. Do not remove desiccant. Protect from moisture. Keep container tightly closed.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients of the following:
New Primary Cutaneous Malignancies
Advise patients to contact their healthcare provider immediately for change in or development of new skin lesions [see Warnings and Precautions (5.1)].
Hemorrhage
Advise patients to notify their healthcare provider immediately with any symptoms suggestive of hemorrhage, such as unusual bleeding [see Warnings and Precautions (5.3)].
Uveitis
Advise patients to contact their healthcare provider if they experience any changes in their vision [see Warnings and Precautions (5.4)].
QT Prolongation
Advise patients that BRAFTOVI can cause QTc interval prolongation and to inform their physician if they have any QTc interval prolongation symptoms, such as syncope [see Warnings and Precautions (5.5)].
Females and Males of Reproductive Potential
Embryo-Fetal Toxicity: Advise females with reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective non-hormonal contraception during treatment with BRAFTOVI and for 2 weeks after the final dose. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with BRAFTOVI [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
Lactation: Advise women not to breastfeed during treatment with BRAFTOVI and for 2 weeks after the final dose [see Use in Specific Populations (8.2)].
Infertility: Advise males of reproductive potential that BRAFTOVI may impair fertility [see Use in Specific Populations (8.3)].
Strong or Moderate CYP3A Inducers or Inhibitors
Coadministration of BRAFTOVI with a strong or moderate CYP3A inhibitor may increase encorafenib concentrations; while coadministration of BRAFTOVI with a strong or moderate CYP3A inducer may decrease encorafenib concentrations. Advise patients that they need to avoid certain medications while taking BRAFTOVI and to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products. Advise patients to avoid grapefruit or grapefruit juice while taking BRAFTOVI [see Drug Interactions (7.1)].
Storage
BRAFTOVI is moisture sensitive. Advise patients to store BRAFTOVI in the original bottle with desiccant and to keep the cap of the bottle tightly closed. Do not remove the desiccants from the bottle.
Distributed by:
Array BioPharma Inc.
3200 Walnut Street
Boulder, CO 80301© 2018 Array BioPharma Inc. All rights reserved.
BRAFTOVI® is a registered trademark of Array BioPharma Inc. in the United States and various other countries.
Patented. See www.arraybiopharma.com/patents



PRINCIPAL DISPLAY PANEL - 75 mg Capsule Bottle Carton - 025-01
NDC 70255-025-01
BRAFTOVI®
(encorafenib) capsules75 mg
Rx only
2 Bottles x 90 Capsules
PRINCIPAL DISPLAY PANEL - 75 mg Capsule Bottle Carton - 025-03
NDC 70255-025-03
BRAFTOVI®
(encorafenib) capsules75 mg
Rx only
2 Bottles x 60 Capsules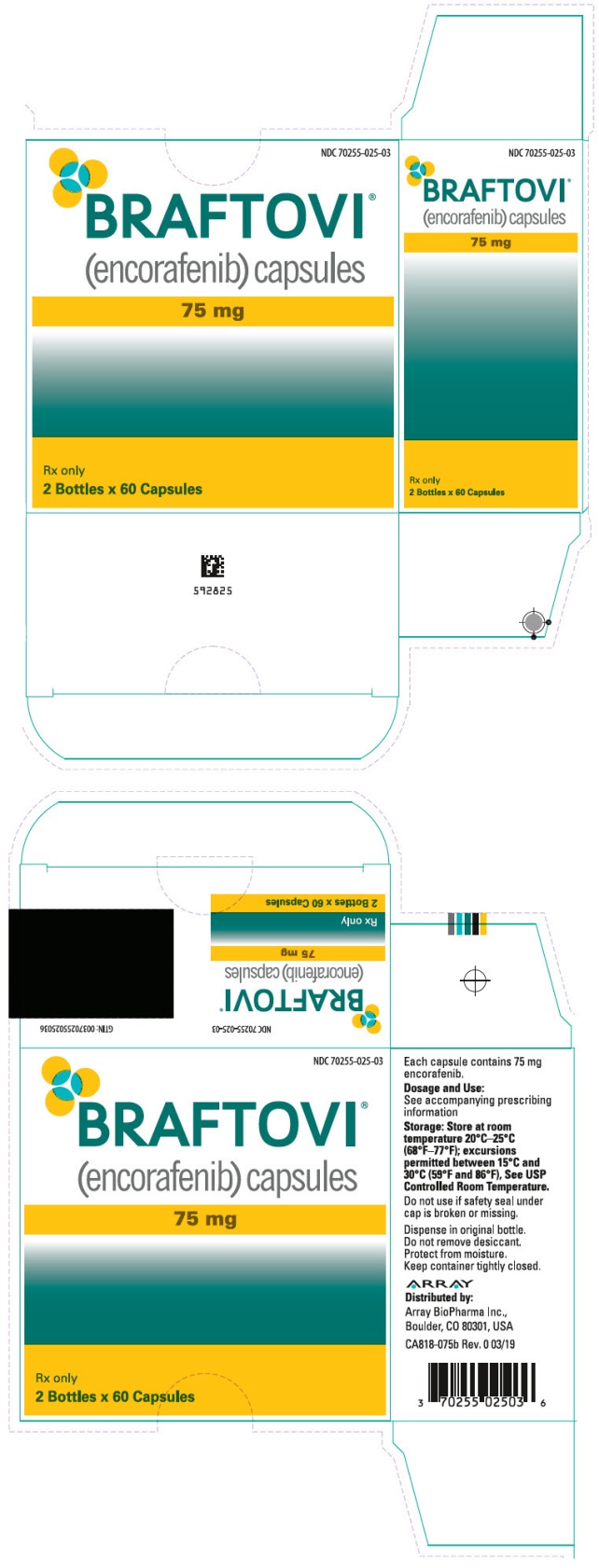
PRINCIPAL DISPLAY PANEL - 50 mg Capsule Bottle Carton
NDC 70255-020-01
BRAFTOVI™
(encorafenib) capsules
50 mg CapsulesRx only
2 Bottles x 60 Capsules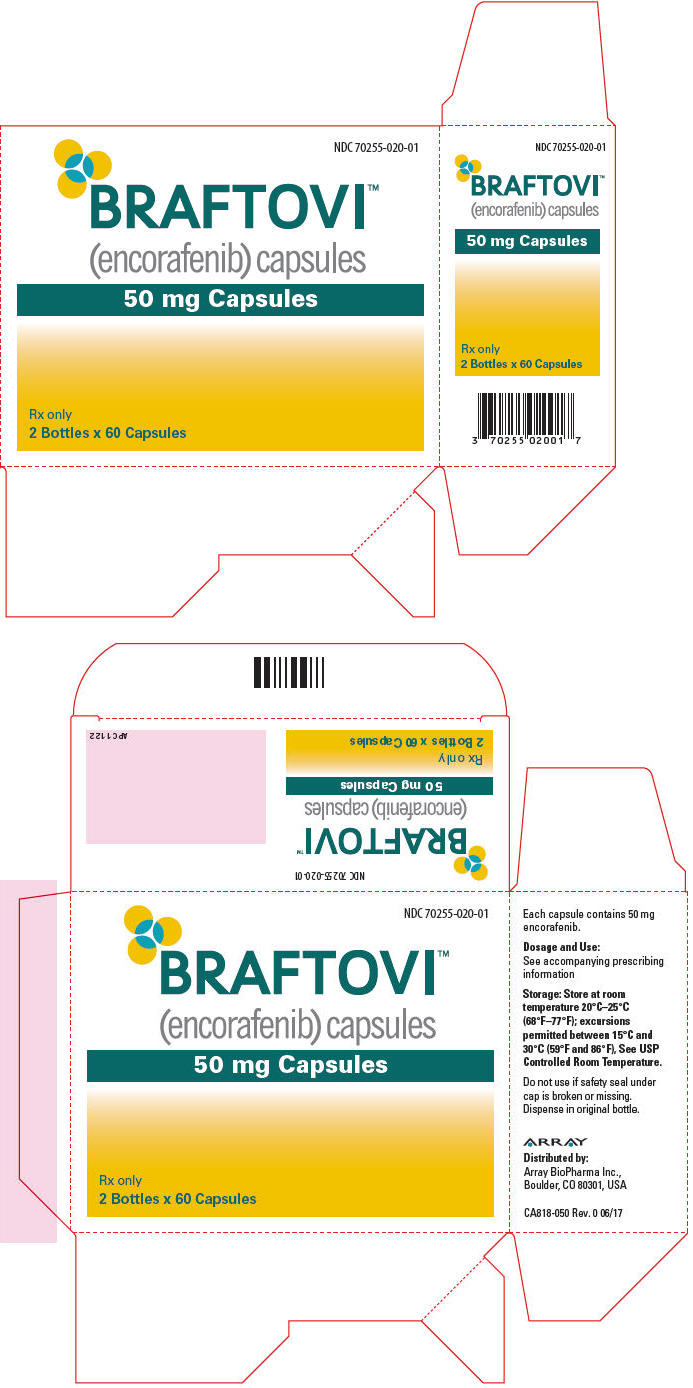
【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。

