商品详情
返回产品目录商品包装及说明书因厂家更换频繁,如有不符以实物为主
洛拉替尼片
国际零售参考价:¥**/瓶
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书
1适应症和用途
LORBRENA ®指示用于其疾病进展的患者的治疗间变性淋巴瘤激酶(ALK)阳性转移性非小细胞肺癌(NSCLC)
克唑替尼和至少一种其他ALK抑制剂用于转移性疾病; 要么
alectinib是首个用于转移性疾病的ALK抑制剂疗法;要么
ceritinib是首个用于转移性疾病的ALK抑制剂疗法。
该适应症是根据肿瘤缓解率和缓解持续时间在加速批准下批准的[见临床研究(14.1) ]。继续批准该适应症可能要取决于验证性试验中对临床益处的验证和描述。
2用法用量
2.1推荐剂量
LORBRENA的推荐剂量为每天 100毫克口服,有或没有食物,直到疾病进展或出现不可接受的毒性[见临床药理学(12.3) ]。
吞咽片整个。请勿咀嚼,压碎或分裂药片。如果片剂破裂,破裂或其他完好无损,请勿摄入。
每天同一时间服用LORBRENA。如果错过了一个剂量,那么除非下次剂量要在4个小时内到期,否则就应服用错过的剂量。不要同时服用2剂,以弥补错过的剂量。
如果LORBRENA后发生呕吐,请不要再服用其他剂量,而应继续下一次计划的剂量。
2.2不良反应的剂量修改
建议减少剂量:
首次剂量减少:每天一次口服LORBRENA 75 mg
第二次减少剂量:每天一次口服LORBRENA 50 mg
无法每天口服一次50 mg的患者,永久停用LORBRENA。
表1 中列出了LORBRENA不良反应的剂量调整方法。
表1 不良反应的推荐LORBRENA剂量修改 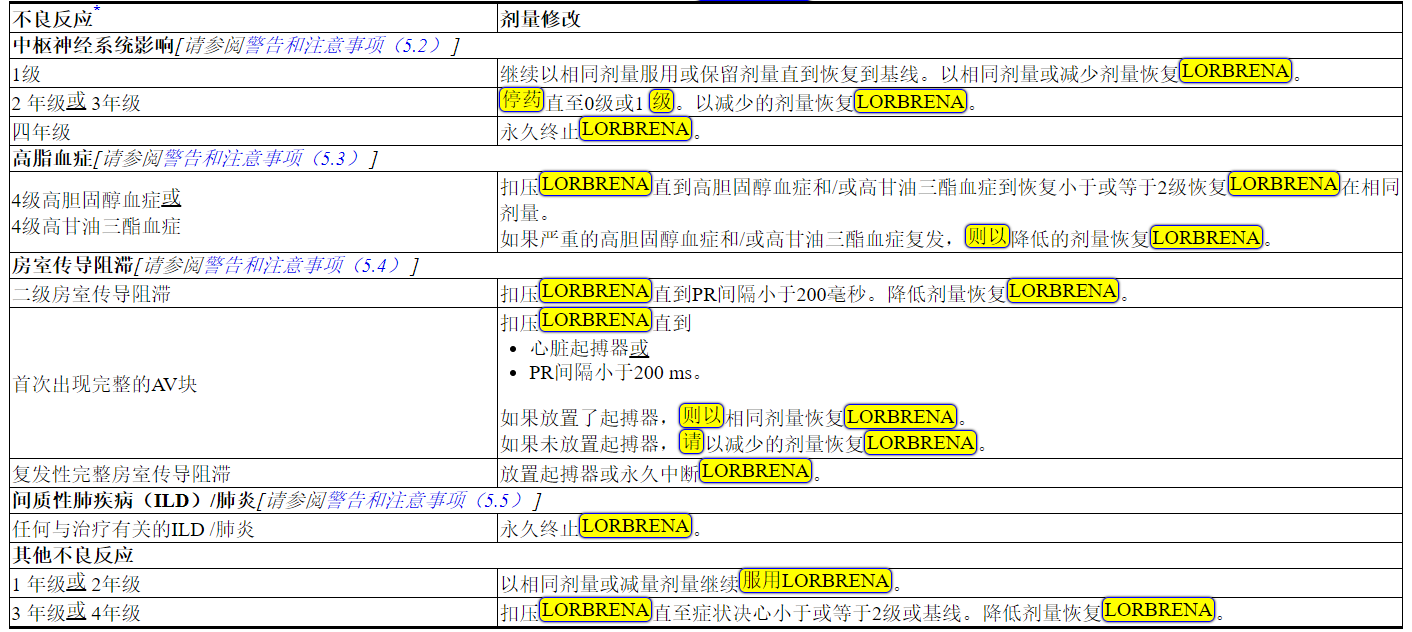
缩写:AV =房室。
*基于美国国家癌症研究所(NCI)不良事件通用术语标准(CTCAE)4.0版的等级。
2.3强效或中效CYP3A诱导剂的同时使用
服用强效CYP3A诱导剂的患者禁用LORBRENA。在开始LORBRENA之前中断强效CYP3A诱导剂的3个血浆CYP3A强效半衰期。避免将LORBRENA与中度CYP3A诱导剂同时使用[见警告和注意事项(5.1),临床药理学(12.3) ]。
2.4强效CYP3A抑制剂的剂量修改
避免将LORBRENA与强效CYP3A抑制剂同时使用。如果不能避免与强效CYP3A抑制剂同时使用,则将LORBRENA的起始剂量从每天一次口服100 mg 减至每天一次口服75 mg。
在因不良反应口服减低剂量至每天一次75 mg且开始使用强效CYP3A抑制剂的患者中,将LORBRENA剂量减低至口服一次每天50 mg。
如果停止同时使用强效CYP3A抑制剂,请将LORBRENA剂量(在强效CYP3A抑制剂的3个血浆半衰期后)增加至开始使用强效抑制剂之前的剂量[见临床药理学(12.3) ]。
3剂型和强度
片剂:
25 mg:8毫米圆形,棕褐色,立即释放,薄膜包衣,在一侧刻有“ Pfizer”,另一侧刻有“ 25”和“ LLN”
100 mg:8.5毫米×17毫米椭圆形,熏衣草,立即释放,薄膜包衣,在一侧压有“ Pfizer”,另一侧压有“ LLN 100”
4禁忌症
服用强效CYP3A诱导剂的患者禁用LORBRENA,因为其可能具有严重的肝毒性[见警告和注意事项(5.1) ]。
5警告和注意事项
5.1并用强效CYP3A诱导剂引起严重肝毒性的风险
接受单剂LORBRENA和每日多次利福平(一种强CYP3A诱导剂)的12名健康受试者中有10名发生严重肝毒性。50%的受试者发生4级丙氨酸氨基转移酶(ALT)或天门冬氨酸氨基转移酶(AST)升高,33%发生3级ALT或AST升高,而8%发生2级ALT或AST升高。ALT或AST升高在3天内发生,中位数15天(7到34天)后又恢复到正常范围内;ALT或AST升高为3或4级的患者,恢复的平均时间为18天; ALT或AST升高为2级的患者,平均恢复时间为7天。
服用强效CYP3A诱导剂的患者禁用LORBRENA。在开始LORBRENA之前中断强效CYP3A诱导剂的3个血浆CYP3A强效半衰期。
避免将LORBRENA与中度CYP3A诱导剂同时使用。如果不能避免伴随使用中等CYP3A诱导的,监视器AST,启动后ALT和胆红素48小时LORBRENA期间启动后的第一周和至少3次LORBRENA。
根据每种药物的相对重要性,中断LORBRENA或CYP3A诱导剂以持续存在2级或更高的肝毒性[见临床药理学(12.3) ]。
5.2中枢神经系统的影响
接受LORBRENA的患者会发生广泛的中枢神经系统(CNS)影响。这些包括癫痫发作,幻觉以及认知功能,情绪(包括自杀意念),言语,精神状态和睡眠的变化。总体而言,中枢神经系统作用发生在接受LORBRENA 的患者中有54%[见不良反应(6.1) ]。332名接受LORBRENA的患者中有29%发生了认知反应研究B7461001中的任何剂量;这些事件中有2.1%是严重的(3或4级)。24%的患者出现情绪影响;这些事件中有1.8%是严重的。14%的患者出现言语影响;这些事件中有0.3%是严重的。幻觉发生在7%的患者中;这些事件中有0.6%是严重的。2.1%的患者发生精神状态改变;这些事件中有1.8%是严重的。3%的患者发生癫痫发作,有时与其他神经系统疾病有关。睡眠影响发生在10%的患者中。首次出现中枢神经系统作用的中位时间为1.2个月(1天至1.7年)。总体而言,有1.5%的患者需要永久停用LORBRENA才能达到中枢神经系统作用;9%的患者需要暂时停药,而8%的患者需要降低剂量。
停药并以相同剂量或减量剂量继续服用,或根据严重程度永久停用LORBRENA [请参阅剂量和给药方法(2.2) ]。
5.3高脂血症
接受LORBRENA的 患者可出现血清胆固醇和甘油三酸酯升高[见不良反应(6.1) ]。在研究B7461001中接受LORBRENA的332例患者中,总胆固醇的3或4级升高发生了17%,甘油三酸酯的3或4级升高发生了。高胆固醇血症和高甘油三酸酯血症的中位发作时间为15天。由于胆固醇和甘油三酯升高,约有7%的患者需要暂时停药,而有3%的患者需要降低LORBRENA的剂量。80%的患者需要开始降脂药物治疗,开始这种药物治疗的中位时间为21天。
在高脂血症患者中开始或增加降脂药的剂量。启动之前监视血清胆固醇和甘油三酯LORBRENA发起后,1 2个月LORBRENA,并在其后定期。首次出现时以相同剂量戒断并恢复;根据严重程度,以相同或减少剂量的LORBRENA进行恢复[见剂量和用法(2.2) ]。
5.4房室传导阻滞
接受LORBRENA的 患者可能发生PR间期延长和房室(AV)阻滞[见不良反应(6.1),临床药理学(12.2) ]。在谁收到295例LORBRENA,剂量为100毫克,每天口服一次在研究B7461001和谁了基线心电图(ECG),1%经历了AV块和0.3%经历3级AV块和后行起搏器放置。
在启动LORBRENA之前和之后定期监测ECG 。对于接受起搏器放置的患者,应减少剂量或以相同剂量戒断并恢复。没有起搏器的患者,请永久终止其复发[参见剂量和给药方法(2.2) ]。
5.5间质性肺疾病/肺炎
LORBRENA可能发生与间质性肺疾病(ILD)/肺炎相一致的严重或危及生命的肺部不良反应。在B7461001研究中,接受任何剂量的LORBRENA的患者中有1.5%发生了ILD /肺炎,其中1.2%的患者患有3级或4级ILD /肺炎。一名患者(0.3%)因ILD /肺炎而中断了LORBRENA的治疗。
对于任何表现出指示ILD /肺炎(例如呼吸困难,咳嗽和发烧)的呼吸道症状加重的患者,应及时调查ILD /肺炎。如果怀疑患有ILD /肺炎,应立即停用LORBRENA。对于任何与治疗相关的ILD /任何严重程度的肺炎,均应永久停用LORBRENA [请参阅剂量和给药方法(2.2) ]。
5.6胚胎-胎儿毒性
根据动物研究的结果及其作用机理,当对孕妇服用时,LORBRENA可能引起胎儿伤害。在每天一次建议剂量为100 mg的母体暴露下,通过口服管饲法对怀孕的大鼠和兔子进行lorlatinib的口服灌胃,会导致畸形,植入后损失增加和流产,而孕产妇的暴露量等于或小于人类暴露量基于曲线下的面积(AUC)。
建议孕妇注意胎儿的潜在危险。建议具有生殖潜力的女性使用有效的非激素避孕方法,因为在用LORBRENA治疗期间以及最终剂量后至少6个月内,LORBRENA可使激素避孕药无效。提醒男性生殖潜在的女性伴侣治疗过程中使用有效的避孕用LORBRENA和最终剂量后3个月[参见药物相互作用(7.2) ,使用在特殊人群中(8.1,8.3) ,非临床毒理学(13.1) ]。
6不良反应
标签上其他地方描述了以下不良反应:
并用强CYP3A诱导剂引起严重肝毒性的风险[见警告和注意事项(5.1) ]
中枢神经系统影响[请参阅警告和注意事项(5.2) ]
高脂血症[请参阅警告和注意事项(5.3) ]
房室传导阻滞[请参阅警告和注意事项(5.4) ]
间质性肺疾病/肺炎[请参阅警告和注意事项(5.5) ]
6.1临床试验经验
由于临床试验是在广泛不同的条件下进行的,因此无法将在某种药物的临床试验中观察到的不良反应率直接与另一种药物的临床试验中观察到的不良反应率进行比较,并且可能无法反映实际中观察到的不良反应率。
警告和注意事项中的数据反映了332名ALK阳性或ROS1阳性,转移性非小细胞肺癌(NSCLC)患者的LORBRENA暴露,该患者参加了多队列,多国,非对照,剂量寻找和活性评估试验(研究B7461001),他以单剂量或分剂量每天10 mg至200 mg的剂量接受LORBRENA。
数据如下所述反映暴露于LORBRENA在295例谁收到ALK阳性或ROS1阳性转移性NSCLC LORBRENA在研究B7461001每日一次口服100毫克。暴露于所述的时间中位数LORBRENA 12.5个月(第1天至35个月)和接收到的52%LORBRENA为≥12个月。患者特征为中位年龄为53岁(19至85岁),年龄≥65岁(18%),女性(58%),白人(49%),亚裔(37%)和东部合作肿瘤小组(ECOG) )效果状态0或1(96%)。
最常见的不良反应(≥20%)为水肿,周围神经病变,认知障碍,呼吸困难,疲劳,体重增加,关节痛,情绪障碍和腹泻。最常见的实验室异常(≥20%)是高胆固醇血症,高甘油三酯血症,贫血,高血糖症,AST增加,白蛋白血症,ALT升高,脂肪酶升高和碱性磷酸酶升高。
295名患者中有32%发生了严重的不良反应。最常见的严重不良反应是肺炎(3.4%),呼吸困难(2.7%),发热(2%),精神状态改变(1.4%)和呼吸衰竭(1.4%)。致命不良反应发生在2.7%的患者中,包括肺炎(0.7%),心肌梗塞(0.7%),急性肺水肿(0.3%),栓塞(0.3%),周围动脉闭塞(0.3%)和呼吸窘迫( 0.3%)。8%的患者永久终止了LORBRENA的不良反应。
导致永久停药的最常见不良反应是呼吸衰竭(1.4%),呼吸困难(0.7%),心肌梗塞(0.7%),认知影响(0.7%)和情绪影响(0.7%)。大约48%的患者需要中断剂量。导致剂量中断的最常见不良反应是水肿(7%),高甘油三酯血症(6%),周围神经病变(5%),认知作用(4.4%),脂肪酶升高(3.7%),高胆固醇血症(3.4%),情绪影响(3.1%),呼吸困难(2.7%),肺炎(2.7%)和高血压(2.0%)。大约有24%的患者需要减少至少1剂不良反应剂量。导致剂量减少的最常见不良反应是水肿(6%),周围神经病变(4.7%),认知作用(4.1%)和情绪影响(3.1%)。
表2和表3分别总结了在研究B7461001中接受LORBRENA治疗的患者的常见不良反应和实验室异常。
表2在研究B7461001中,≥10%的患者发生不良反应*

缩写:NCI CTCAE =美国国家癌症研究所不良事件通用术语标准;SOC =系统器官类别。
*不良反应使用NCI CTCAE 4.0版进行分级。
†情绪影响(包括情绪障碍,影响不稳定,攻击性,躁动,焦虑,沮丧的情绪,抑郁,欣快的情绪,易怒,躁狂,情绪改变,情绪波动,性格改变,压力,自杀意念)。
‡周围神经病变(包括烧灼感,腕管综合症,感觉异常,下肢,步态障碍,感觉不足,肌肉无力,神经痛,周围神经病变,神经毒性,感觉异常,周围感觉神经病变,感觉障碍)。
§认知影响(包括来自SOC神经系统疾病的事件:健忘症,认知障碍,痴呆,注意力障碍,记忆障碍,精神障碍;还包括来自SOC精神病的事件:注意缺陷/多动障碍,精神错乱,del妄,神志不清,阅读障碍)。
¶言语影响(包括失语症,构音障碍,言语缓慢,言语障碍)
#睡眠影响(包括异常的梦,失眠,噩梦,睡眠障碍,睡眠障碍,梦游)
Þ视力障碍(包括失明,复视,畏光,验光,视力模糊,视力下降,视力障碍,玻璃体漂浮物)。
ß肌痛(包括肌肉骨骼疼痛,肌痛)。
一种水肿(包括水肿,周围性水肿,眼睑水肿,面部水肿,全身性水肿,局部性水肿,眶周水肿,周围肿胀,肿胀)。
è疲劳(包括乏力,疲劳)。
ð上呼吸道感染(包括真菌上呼吸道感染,上呼吸道感染,病毒性上呼吸道感染)。
ø皮疹(包括痤疮性皮炎,斑丘疹,瘙痒性皮疹,皮疹)。
幻觉(7%)发生在1%到10%之间的其他临床上显着的不良反应。
表3在研究B7461001中,≥20%的患者发生的实验室值变差*
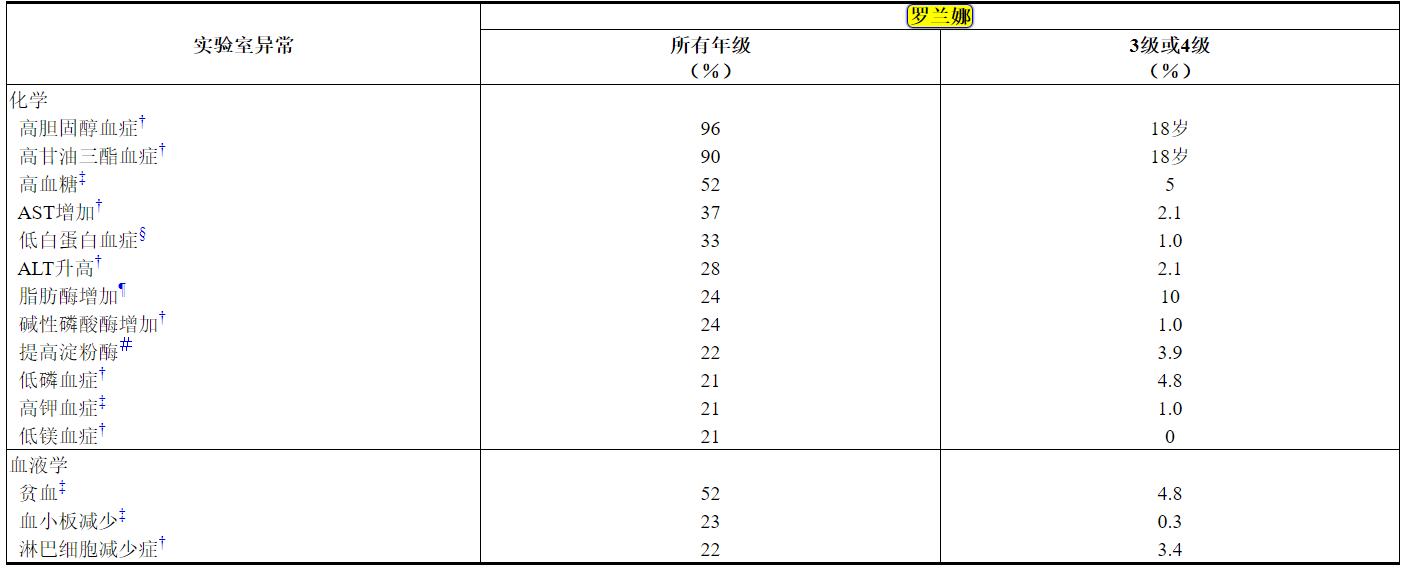
缩写:ALT =丙氨酸转氨酶;AST =天冬氨酸转氨酶;NCI CTCAE =美国国家癌症研究所不良事件通用术语标准。
N =至少对一项感兴趣参数进行了研究评估的患者人数。*使用NCI CTCAE 4.0版进行评分。
†N = 292。
‡N = 293。
§N = 291。
¶N = 290。
#N = 284。
7药物相互作用
7.1其他药物对LORBRENA的影响
CYP3A诱导剂的作用
将LORBRENA与强效CYP3A诱导剂同时使用会降低lorlatinib血浆浓度,这可能会降低LORBRENA的疗效。尚未研究将LORBRENA与中度CYP3A诱导剂同时使用对lorlatinib血浆浓度的影响。
在接受LORBRENA并使用利福平(一种强CYP3A诱导剂)的健康受试者中发生严重的肝毒性。在接受单次100毫克剂量的LORBRENA和每日多次利福平的12名健康受试者中,83%的受试者ALT或AST升高3或4级,而ALT或AST的2级升高则发生8%。肝毒性的可能机制是通过LORBRENA和rifampin(两者都是PXR激动剂)激活孕烷X受体(PXR)。并用LORBRENA和中度CYP3A诱导剂(也是PXR激动剂)的肝毒性风险未知。
服用强效CYP3A诱导剂的患者禁用LORBRENA。在开始LORBRENA之前中断强效CYP3A诱导剂的3个血浆CYP3A强效半衰期。
避免将LORBRENA与中度CYP3A诱导剂同时使用。如果不能避免同时使用中度CYP3A诱导剂,则按照推荐的方法监测ALT,AST和胆红素[见剂量和给药方法(2.3),警告和注意事项(5.1),临床药理学(12.3) ]。
强效CYP3A抑制剂的作用
与强效CYP3A抑制剂同时使用会增加lorlatinib的血浆浓度,这可能会增加LORBRENA不良反应的发生率和严重程度。避免将LORBRENA与强效CYP3A抑制剂同时使用。如果不能避免同时使用,请按照推荐的剂量减少LORBRENA的剂量[见剂量和用法(2.4),临床药理学(12.3) ]。
7.2 LORBRENA对其他药物的影响
CYP3A底物
并用LORBRENA会降低CYP3A底物的浓度[见临床药理学(12.3) ],这可能会降低这些底物的功效。避免将LORBRENA与CYP3A底物同时使用,因为最小的浓度变化可能会导致严重的治疗失败。如果不可避免的同时使用,根据批准的产品标签增加CYP3A底物的剂量。
8在特定人群中的使用
8.1怀孕
风险摘要
根据动物研究的结果及其作用机理[请参见临床药理学(12.1) ],当将LORBRENA施用给孕妇时,可能对胚胎造成胎儿伤害。目前尚无孕妇使用洛比娜的数据。在每天一次建议剂量为100 mg的母体暴露下,通过口服管饲法对怀孕的大鼠和兔子进行lorlatinib的口服灌胃,会导致畸形,植入后损失增加和流产,而孕产妇的暴露量等于或小于人类暴露量基于AUC (请参阅数据)。建议孕妇注意胎儿的潜在危险。
在美国普通人群中,临床公认的怀孕中主要先天缺陷和流产的估计背景风险分别为2%至4%和15%至20%。
数据
动物资料
在大鼠和兔子中进行了初步的胚胎-胎儿发育研究,研究了在器官生成期间服用洛来替尼的情况。在兔子中,以15 mg / kg的剂量(建议使用100 mg剂量的人体暴露量的3倍左右)使用lorlatinib会导致流产和完全丧失妊娠。以4 mg / kg的剂量(约为100 mg推荐剂量的人暴露量的0.6倍),毒性包括植入后损失增加和畸形,包括四肢旋转,肾脏畸形,头顶圆顶,上arch弓和扩张。脑室。在大鼠中,以4 mg / kg的剂量服用lorlatinib会导致完全丧失妊娠(在100 mg的推荐剂量下约为人暴露的5倍)。
8.2哺乳
风险摘要
没有关于人乳或动物乳中氯雷替尼或其代谢物的存在或其对母乳喂养婴儿或产奶量的影响的数据。由于母乳喂养的婴儿可能会出现严重的不良反应,因此请指示妇女在接受LORBRENA治疗期间以及最终剂量后的7天内不要母乳喂养。
8.3生殖潜力的男性和女性
验孕
在开始LORBRENA 之前,验证具有生殖潜能的女性的怀孕状况[请参阅在特定人群中使用(8.1) ]。
避孕
当给予孕妇时,洛伯那能引起胚胎-胎儿的伤害[见特定人群的使用(8.1) ]。
女性
建议有生殖潜力的女性患者在接受LORBRENA治疗期间以及最终剂量后至少6个月内使用有效的非激素避孕方法。建议有生殖能力的女性使用非激素的避孕方法,因为LORBRENA可以使激素避孕药失效[见药物相互作用(7.2) ]。
雄性
根据遗传毒性结果,建议具有生殖潜能的女性伴侣的男性在接受LORBRENA治疗期间以及最终剂量后至少3个月内使用有效的避孕药[请参见非临床毒理学(13.1) ]。
不孕症
雄性
根据动物研究的结果,LORBRENA可能会暂时损害男性的生育能力[请参阅非临床毒理学(13.1) ]。
8.4小儿使用
尚未确定LORBRENA在小儿患者中的安全性和有效性。
8.5老年人使用
在研究B7461001中的295位患者中,他们每天口服一次100 mg的LORBRENA,其中18%的患者年龄在65岁以上。尽管数据有限,但在65岁以上的患者和较年轻与较年轻的患者之间,在安全性或疗效方面没有观察到临床上的重要差异。
8.6肝功能不全
对于轻度肝功能不全(AST≥ULN的总胆红素≤正常[ULN]上限或任何AST的总胆红素> 1到1.5×ULN的患者),建议不调整剂量。对于中度或重度肝功能不全的患者,尚未确定LORBRENA的推荐剂量[请参见临床药理学(12.3) ]。
8.7肾功能不全
对于轻度或中度肾功能不全的患者(Cockcroft-Gault估计肌酐清除率[CLcr]为30至89 mL / min),建议不调整剂量。对于严重肾功能不全的患者,尚未确定LORBRENA的推荐剂量[请参见临床药理学(12.3) ]。
11说明
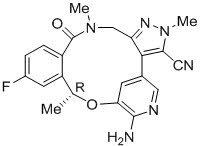
LORBRENA(lorlatinib)是口服的激酶抑制剂。分子式为C 21 H 19 FN 6 O 2(无水形式),分子量为406.41道尔顿。化学名称为(10 R)-7-氨基-12-氟-2,10,16-三甲基-15-氧代10,15,16,17-四氢-2 H -4,8-甲并吡唑[4, 3- ħ ] [2,5,11]苯并氧-3-腈。化学结构如下所示:
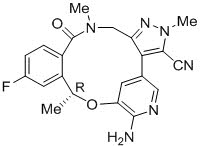
罗拉替尼是白色至类白色粉末,pKa为4.92。氯雷替尼在水性介质中的溶解度在pH 2.55至pH 8.02范围内从32.38 mg / mL降低至0.17 mg / mL。pH值为9时,分配系数(辛醇/水)的对数为2.45。
LORBRENA以包含25 mg或100 mg lorlatinib的片剂形式提供,其中含有以下非活性成分:微晶纤维素,无水磷酸氢钙,羟乙酸淀粉钠和硬脂酸镁。薄膜包衣包含羟丙基甲基纤维素(HPMC)2910 /羟丙甲纤维素,乳糖一水合物,聚乙二醇/聚乙二醇(PEG)3350,三醋精,二氧化钛,二氧化亚铁/氧化铁黑和氧化铁红。
12临床药理学
12.1行动机制
Lorlatinib是一种激酶抑制剂,对ALK和ROS1以及TYK1,FER,FPS,TRKA,TRKB,TRKC,FAK,FAK2和ACK具有体外活性。洛拉替尼显示出对多种突变形式的ALK酶的体外活性,包括在克唑替尼和其他ALK抑制剂的疾病进展时在肿瘤中检测到的某些突变。
在皮下植入了带有带有ALK变体1或ALK突变的EML4融合体的肿瘤的小鼠,包括在疾病进展时在ALK抑制剂上在肿瘤中检测到的G1202R和I1171T突变,施用lorlatinib会产生抗肿瘤活性。罗拉替尼还显示出在颅内植入EML4-ALK驱动的肿瘤细胞系的小鼠中的抗肿瘤活性和延长的生存期。氯雷替尼在体内模型中的总体抗肿瘤活性是剂量依赖性的,并且与ALK磷酸化的抑制作用有关。
12.2药效学
暴露-反应关系
根据研究B7461001的数据,在推荐剂量下达到稳态暴露时,观察到3级或4级高胆固醇血症以及任何3级或4级不良反应的暴露-反应关系,且发生以下不良反应的可能性更高:增加洛来替尼暴露。
心脏电生理学
在研究B7461001中,每天接受100 mg推荐剂量的LORBRENA并接受心电图测量的295位患者中,PR区间相对于基线的最大平均变化为16.4 ms(2面90%上置信度区间[CI] 19.4 ms )。在284名基线PR间隔<200 ms的患者中,有14%的患者在开始LORBRENA后PR间隔延长≥200ms。PR间隔的延长以浓度依赖性方式发生。1%的患者发生房室传导阻滞。
在研究B7461001的活动估计部分中以推荐剂量接受LORBRENA的 275名患者中,未检测到QTcF间隔(即> 20 ms)相对于基线的大平均值增加。
12.3药代动力学
在每天一次口服10 mg至200 mg的剂量范围内,稳态lorlatinib的最大血浆浓度(C max)成比例增加,而AUC的增加略小于成比例的增加(建议剂量的0.1至2倍)。在推荐剂量下,癌症患者的平均C max(变异系数[CV]%)为577 ng / mL(42%),AUC 0–24h为5650 ng∙h / mL(39%)。与单剂量相比,洛拉替尼的口服清除率在稳态下增加,表明具有自诱导作用。
吸收性
单次口服100 mg剂量后,洛拉替尼T max的中位数为稳定状态下单次口服100 mg剂量后为1.2小时(0.5至4小时),口服100 mg每天一次后为2小时(0.5至23小时)。
与静脉内给药相比,口服后平均绝对生物利用度为81%(90%CI为75.7%,86.2%)。
食物的作用
给药LORBRENA具有高脂肪,高热量膳食(大约1000个卡路里来自蛋白质150个卡路里,碳水化合物250个卡路里,以及从脂肪500至600卡路里)对lorlatinib药代动力学没有临床上有意义的效果。
分配
在体外,洛拉替尼以2.4 µM的浓度与血浆蛋白结合率为66%。血液与血浆的比率为0.99。分布(V的平均值(CV%)稳态体积SS)是在单次静脉内剂量305 L(28%)。
消除
平均血浆半衰期(T ½ lorlatinib的)是单次口服100毫克的剂量后24小时(40%)LORBRENA。单次口服100 mg剂量后,平均口服清除率(CL / F)为11 L / h(35%),稳定状态下增加至18 L / h(39%),表明自诱导。
代谢
在体外,lorlatinib主要由CYP3A4和UGT1A4代谢,而CYP2C8,CYP2C19,CYP3A5和UGT1A3的贡献较小。
在血浆中,洛拉替尼的酰胺和芳香醚键的氧化裂解产生的洛拉替尼的苯甲酸代谢物(M8)占人体[ 14 C]质量平衡研究中循环放射性的21%。氧化裂解代谢产物M8在药理上是无活性的。
排泄
单次口服100 mg放射性标记的lorlatinib后,尿液中回收了48%的放射性(<1%不变),粪便中回收了41%(约9%不变)。
特定人群
根据年龄(19至85岁),性别,种族/族裔,体重,轻至中度肾功能不全(CLcr 30至89 mL / min),轻度肝功能不全(总胆红素≤),未观察到lorlatinib药代动力学的临床意义差异。 ULN和AST> ULN或总胆红素> 1.5×ULN和任何AST),或CYP3A5和CYP2C19的代谢物表型。中度至重度肝功能不全或重度肾损伤的药代动力学lorlatinib的效果是未知的[见特殊人群中使用(8.6,8.7) ]。
药物相互作用研究
临床研究
CYP3A诱导剂对Lorlatinib的影响: 12名健康受试者接受利福平(一种也能激活PXR的强CYP3A诱导剂),600 mg每天一次,连续8天(第1至8天),并在第8天单次口服100 mg剂量的LORBRENA。利福平与LORBRENA联用可将lorlatinib AUC inf平均降低85%,C max减少了76%在3天内发生ALT或AST的2至4级升高。受试者的4级ALT或AST升高发生在50%,三级ALT或AST升高发生在33%,2级ALT或AST升高发生在8%的受试者中。ALT和AST在7到34天(中位数为15天)内恢复到正常范围内。尚不知道同时使用中度CYP3A诱导剂对lorlatinib药代动力学的影响或肝毒性风险以及同时使用中度CYP3A诱导剂的作用[见药物相互作用(7.1) ]。
上Lorlatinib斯特朗CYP3A抑制剂的效果:伊曲康唑,强大的CYP3A抑制剂,增加的AUC INF了42%和增加的C ^ 最大由24%单次口服100毫克剂量的LORBRENA [见药物相互作用(7.1) ]。
洛拉替尼对CYP3A底物的影响: 单次口服2 mg咪达唑仑(一种敏感的CYP3A底物),每天口服150 mg 罗布瑞娜,持续15天,AUC inf降低64%,Cmax降低50%[参见药物相互作用(7.2) ]。
减酸剂对Lorlatinib的影响:质子泵抑制剂雷贝拉唑的同时使用对lorlatinib的药代动力学没有临床意义的影响。
体外研究
Lorlatinib对CYP酶的影响:体外研究表明lorlatinib是CYP3A的时间依赖性抑制剂和诱导剂,并且它激活PXR,体内的净作用是诱导。罗拉替尼诱导CYP2B6并激活人类组成型雄激素受体(CAR)。洛拉替尼和主要循环代谢物M8不抑制CYP1A2,CYP2B6,CYP2C8,CYP2C9,CYP2C19和CYP2D6。M8不抑制CYP3A。
M8不诱导CYP1A2,CYP2B6和CYP3A。
罗拉替尼对UDP-葡萄糖醛酸转移酶(UGT)的影响:罗拉替尼和M8不会抑制UGT1A1,UGT1A4,UGT1A6,UGT1A9,UGT2B7和UGT2B15。
洛拉替尼对转运蛋白的影响:洛拉替尼抑制P-糖蛋白(P-gp),有机阳离子转运蛋白(OCT)1,有机阴离子转运蛋白(OAT)3,多药和毒素挤出(MATE)1和肠癌抗性蛋白(BCRP) )。罗拉替尼不抑制有机阴离子转运多肽(OATP)1B1,OATP1B3,OAT1,OCT2,MATE2K和系统性BCRP。M8不抑制P-gp,BCRP,OATP1B1,OATP1B3,OAT1,OAT3,OCT1,OCT2,MATE1和MATE2K。
13毒理学
13.1致癌,诱变,生育力受损
尚未对lorlatinib进行致癌性研究。洛拉替尼在人淋巴母细胞TK6细胞的体外测定中具有成瘤作用,并且在大鼠骨髓中体内微核形成呈阳性。在体外细菌反向突变(Ames)分析中,洛拉替尼不具有致突变性。
没有使用lorlatinib进行专门的生育力研究。男性生殖器官的发现发生在重复剂量毒性研究中,包括较低的睾丸重量,附睾重量和前列腺重量。睾丸小管变性/萎缩;前列腺萎缩 和/或附睾炎症,分别在大鼠和狗中分别为15 mg / kg /天和7 mg / kg /天(以100毫克的推荐剂量(基于AUC),分别为人体暴露量的8倍和2倍)。对男性生殖器官的影响是可逆的。
13.2动物毒理学和/或药理学
动物腹部腹部伸张,皮疹,胆固醇和甘油三酸酯升高。这些发现伴随着大鼠15 mg / kg / day和狗2 mg / kg / day肝脏的增生和胆管扩张以及胰脏腺泡萎缩(分别是8倍和0.5倍)。以100毫克的推荐剂量(基于AUC)进行人体暴露)。在恢复期内,所有影响都是可逆的。
14临床研究
14.1先前用ALK激酶抑制剂治疗过的ALK阳性转移性NSCLC
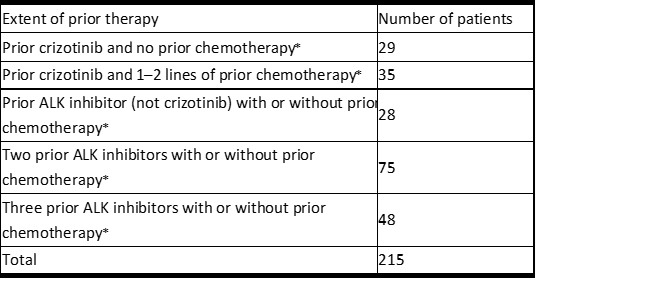
LORBRENA的功效在先前用一种或多种ALK激酶抑制剂治疗的ALK阳性转移性非小细胞肺癌(NSCLC)患者亚组中进行了研究,这些患者参加了非随机,剂量范围和活性估计,多队列,多中心研究(研究B7461001; NCT01970865)。根据实体肿瘤反应评估标准(RECIST)1.1版(v1.1),ECOG表现状态为0至2,并记录了ALK重排,该亚组中的患者必须患有转移性疾病,且其靶标病灶至少为1个。通过荧光原位杂交(FISH)分析或免疫组织化学(IHC)确定的肿瘤组织,并接受LORBRENA每天一次口服100毫克。入选前2周内无症状CNS转移的患者(包括稳定使用或减少类固醇使用的患者)是合格的。患有严重,急性或慢性精神病(包括自杀意念或行为)的患者被排除在外。另外,对于ALK阳性转移性NSCLC患者,为每个单独的队列指定了先前治疗的程度和类型(见表4)。根据RECIST v1.1,主要疗效结局指标为总体缓解率(ORR)和颅内ORR,由独立中央审查(ICR)委员会评估。数据汇总于表4中列出的所有子组中。其他疗效结果指标包括反应持续时间(DOR)和颅内DOR。
表4中的亚组共计215名患者。表4提供了按既往治疗类型和程度划分的患者分布。所有215名患者的人口统计学特征是:女性59%,白人51%,白人34%亚洲人,年龄中位数为53岁(29至85岁),其中18%≥65岁的患者。96%的患者在基线时的ECOG表现状态为0或1。所有患者均患有转移性疾病,95%患有腺癌。ICR鉴定的脑转移发生在69%的患者中。其中,有60%的患者先前接受过脑部放疗,而每个ICR的患者中有60%(n = 89)有可测量的疾病。

缩写:ALK =间变性淋巴瘤激酶;NSCLC =非小细胞肺癌。
*在转移环境中进行化学疗法。
表5和表6汇总了研究B7461001的功效结果。
表5研究B7461001中的功效结果 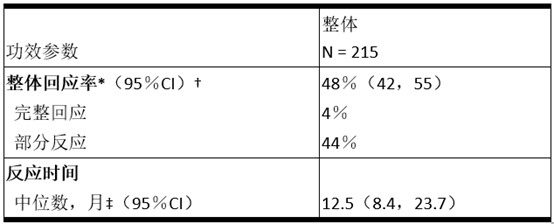
缩写:CI =置信区间;N =患者数。
*根据独立的中央审查。
†使用基于二项式分布的精确方法。
‡使用Kaplan Meier方法估算。
表6汇总了根据RECIST v1.1对研究B7461001中具有基线可测量病变的89例患者亚组的颅内ORR和对CNS转移反应持续时间的评估。其中,有56例(63%)患者接受过先前的脑部放射治疗,包括42名患者(47%)在开始使用LORBRENA治疗之前至少6个月完成了脑部放射治疗。
表6研究B7461001中可测量的颅内病变患者的颅内反应率 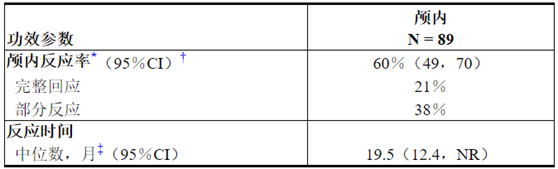
缩写:CI =置信区间;N =患者人数;NR =未达到。
*根据独立的中央审查。
†使用基于二项式分布的精确方法。
‡使用Kaplan-Meier方法估算。
在先前治疗定义的亚组中进行的探索性分析中,对LORBRENA的缓解率是:
- 119名接受克唑替尼和至少一种其他ALK抑制剂并接受或未接受化疗的患者的ORR = 39%(95%CI:30,48)
- 在接受或不接受化疗的13例患者中,将alectinib作为其唯一的ALK抑制剂,ORR = 31%(95%CI:9,61)
- 在接受或不接受化疗的13例接受塞来替尼作为其唯一ALK抑制剂的患者中,ORR = 46%(95%CI:19,75)
16供应/存储和处理方式
表7描述了LORBRENA的可用优势和包装配置:
表7 LORBRENA片剂 
存放在20°C至25°C(68°F至77°F); 允许的偏移范围是15°C至30°C(59°F至86°F)[请参阅USP控制的室温]。
17患者咨询信息
建议患者阅读FDA批准的患者标签(患者信息)。
并用强效CYP3A诱导剂引起严重肝毒性的风险
同时使用强效CYP3A诱导剂告知患者肝毒性的潜在风险。
建议患者将他们正在服用的所有药物告知其医疗保健提供者,包括处方药,非处方药,维生素和草药产品(例如,圣约翰草)[请参阅警告和注意事项(5.1) ]。
中枢神经系统(CNS)的影响
如果患者出现新的或恶化的中枢神经系统症状,建议患者通知其医疗服务提供者[请参阅警告和注意事项(5.2) ]。
高脂血症
告知患者在治疗期间将监测血清胆固醇和甘油三酸酯。告知患者可能需要开始或增加降脂药的剂量[见警告和注意事项(5.3) ]。
房室传导阻滞
告知患者房室传导阻滞的风险。劝告患者立即联系他们的医疗保健提供者以报告新的或恶化的心脏症状[见警告和注意事项(5.4) ]。
间质性肺疾病(ILD)/肺炎
告知患者严重ILD /肺炎的风险。建议患者立即联系他们的医疗保健提供者,以报告新的或恶化的呼吸道症状[见警告和注意事项(5.5) ]。
胚胎-胎儿毒性
告知女性生殖潜能对胎儿的潜在危险。劝告女性告知其已知或怀疑怀孕的医疗服务提供者[请参阅警告和注意事项(5.6),在特定人群中使用(8.1) ]。
建议有生殖潜力的女性在接受LORBRENA治疗期间以及最终剂量后至少6个月内使用有效的非荷尔蒙避孕药(请参见“ 在特定人群中使用(8.3) ”)。
建议具有生殖潜力的女性伴侣的男性患者在接受LORBRENA治疗期间以及最终剂量后至少3个月内使用有效的避孕药[请参见“ 在特殊人群中使用(8.3),非临床毒理学(13.1) ”。
哺乳期
劝告妇女在用LORBRENA治疗期间和最终剂量后7天内不要母乳喂养[见在特定人群中使用(8.2) ]。
不孕症
告知有生殖潜力的男性,LORBRENA可能会暂时损害生育能力[请参见“ 在特定人群中使用(8.3),非临床毒理学(13.1) ”。
该产品的标签可能已更新。有关完整的处方信息,请访问www。LORBRENA.com。

LAB-1162-1.0
病人资料
LORBRENA(lor-BREN-ah)
(lorlatinib)
片我应该了解的关于LORBRENA的最重要信息是什么?
LORBRENA可能引起严重的副作用,包括:
- 与其他药物相互作用引起的肝脏问题。重要的是要知道LORBRENA不应服用哪些药物。
- 脑部问题(中枢神经系统[CNS])起作用。许多患者的脑功能出现问题,包括思维(如健忘或困惑),情绪(如抑郁),言语,看到或听到不真实的事物(晕眩)和在用LORBRENA治疗期间出现癫痫发作。在某些患者中,这些问题很严重,您的医疗服务提供者可能需要停止服用洛贝那。
- 增加血液中胆固醇和甘油三酸酯(脂质)的水平。在用LORBRENA治疗期间,大多数患者的血脂水平会增加。
- 如果您在用LORBRENA治疗期间血液中的脂质水平增加,则您的医疗保健提供者可能需要开始服用降低其水平的药物。如果您已经在服用某种药物来降低血液中的脂质水平,则您的医疗保健提供者可能需要增加该药物的剂量。
- 您的医疗保健提供者应在开始治疗之前,开始治疗后1至2个月以及在用LORBRENA治疗期间进行血液检查,以检查血液中的脂质水平。
- 心脏问题。 LORBRENA可能会导致心跳缓慢或异常。您的医护人员应在开始使用LORBRENA之前和治疗期间检查您的心律(心电图[EKG])。如果您感到头晕或晕眩或心跳异常,请立即告诉您的医疗保健提供者。在某些患者中,这些问题很严重,您的医疗服务提供者可能需要停止服用洛美那或放置起搏器。
- 肺部问题。 LORBRENA可能在治疗期间引起严重的或危及生命的肺部肿胀(发炎),并可能导致死亡。症状可能与肺癌相似。如果您有任何新的或恶化的肺部症状,包括呼吸困难,呼吸急促,咳嗽或发烧,请立即告知您的医疗服务提供者。
在某些患者中,这些问题很严重,您的医疗服务提供者可能需要停止服用洛贝那。有关副作用的更多信息,请参见“ LORBRENA的可能的副作用是什么? ”。
什么是LORBRENA?
LORBRENA是一种处方药,用于治疗非小细胞肺癌(NSCLC)的人
- 是由异常的间变性淋巴瘤激酶(ALK)基因引起的,并且
- 已经扩散到您身体的其他部位,
- 曾服用alectinib或ceritinib药物,或已服用crizotinib和至少一种其他药物来治疗由ALK基因引起的NSCLC的患者,以及
- 他们的非小细胞肺癌不再对这些治疗产生反应。
尚不知道LORBRENA在儿童中是否安全有效。
如果您服用某些称为强CYP3A诱导剂的药物,请勿服用LORBRENA。如果您不确定,请向您的医疗保健提供者提供这些药物的清单。
服用LORBRENA之前,请告知您的医疗保健提供者您的所有医疗状况,包括是否:
- 正在服用其他药物
- 曾有过抑郁或癫痫发作
- 血液中胆固醇或甘油三酸酯含量高
- 心跳有问题
- 有肺或呼吸问题
- 正在怀孕或打算怀孕。罗兰娜会伤害未出生的婴儿。
- 您的医疗保健提供者将在您开始使用LORBRENA治疗之前进行妊娠试验。
- 如果您怀孕或认为您在用LORBRENA治疗期间可能怀孕,请立即告诉您的医疗服务提供者。
–
能够怀孕的女性应在LORBRENA治疗期间以及LORBRENA最终剂量后至少6个月内使用有效的非激素避孕措施。如果在LORBRENA治疗期间使用避孕药(口服避孕药)和其他激素控制型避孕药可能无效。与您的医疗保健提供者讨论在此期间适合您的节育选择。
–
拥有女性伴侣且能够怀孕的男性,在LORBRENA治疗期间和最终LORBRENA给药后至少3个月内应使用有效的避孕方法。
- 正在母乳喂养或计划母乳喂养。不知道LORBRENA是否会进入母乳。在用洛布瑞娜治疗期间以及最终剂量后的7天内,请勿母乳喂养。与您的医疗保健提供者谈谈这段时间内喂养婴儿的最佳方法。
告诉您的医疗保健提供者您服用的所有药物,包括处方药,非处方药,维生素和草药补品。
我应该如何服用洛比娜?
- 完全按照医护人员的指示服用洛贝那。除非您的医疗保健提供者告诉您,否则请勿更改剂量或停止服用洛贝那。
- 如果您出现副作用,您的医疗保健提供者可能会更改您的剂量,暂时停止或永久停止LORBRENA的治疗。
- 吞下整个LORBRENA片。请勿咀嚼,压碎或分裂LORBRENA片剂。不要服用LORBRENA片剂,如果它们破裂,破裂或不完整。
- 每天大约在同一时间服用LORBRENA。
- 您可以在有或没有食物的情况下服用LORBRENA。
- 如果您错过了剂量,请记得记住。但是,如果接近您的下一次服药时间(4小时内),则只需在常规时间服用下一次服药即可。
- 如果您在服用LORBRENA后呕吐,请不要再服用。在正常时间服用下一次剂量。
LORBRENA可能有哪些副作用?
- 请参阅“ 关于LORBRENA,我应该了解的最重要的信息是什么? ”
LORBRENA最常见的副作用包括:
- 手臂,腿,手和脚肿胀(浮肿)
- 关节或手臂和腿部麻木和刺痛感(周围神经病变)
- 思维困难或困惑
- 呼吸困难
- 疲劳(疲劳)
- 体重增加
- 关节疼痛
- 情绪变化,悲伤或焦虑
- 腹泻
LORBRENA可能导致男性生育力下降。在男性中,这可能会影响您生孩子的能力。如果您担心生育,请与您的医疗保健提供者联系。
这些并不是LORBRENA的所有可能的副作用。有关更多信息,请咨询您的医疗保健提供者或药剂师。
致电您的医疗保健提供者以获取有关副作用的医疗建议。您可以通过1-800-FDA-1088向FDA报告副作用。
我应该如何存放洛伦娜?
- 将LORBRENA储存在68°F至77°F(20°C至25°C)的室温下。
请勿将LORBRENA和所有药物放在儿童接触不到的地方。
有关安全有效使用LORBRENA的一般信息
有时出于患者信息手册中列出的目的以外的目的开出处方药。请勿在没有规定的条件下使用LORBRENA。即使他人有与您相同的症状,也请勿将LORBRENA给予他人。可能会伤害他们。您可以要求您的医疗保健提供者或药剂师提供有关LORBRENA的更多信息,这些信息是为卫生专业人员编写的。
LORBRENA中的成分是什么?
有效成分:氯雷替尼
非活性成分:微晶纤维素,无水磷酸氢钙,羟乙酸淀粉钠和硬脂酸镁。
薄膜包衣包含:羟丙基甲基纤维素(HPMC)2910 /羟丙甲纤维素,乳糖一水合物,聚乙二醇/聚乙二醇(PEG)3350,三醋精,二氧化钛,四氧化三铁/黑色氧化铁和氧化铁红。
有关更多信息,请访问www。LORBRENA.com。

LAB-1163-1.0
该患者信息已获得美国食品和药物管理局的批准。 2018年11月
主要片剂-25毫克片剂瓶标签
辉瑞
NDC 0069-0227-01
Lorbrena ®
(lorlatinib)片剂
25mg的
仅30 Tablet Rx
主要显示面板-100毫克片剂瓶标签
辉瑞
NDC 0069-0231-01
Lorbrena ®
(lorlatinib)片剂100毫克
仅30 Tablet Rx
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书
-
本说明书来源于FDA网站https://nctr-crs.fda.gov/fdalabel/services/spl/set-ids/2b34d62d-e02a-4af3-bc0d-1571dd4ee76d/spl-doc?hl=LORBRENA
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书
1 INDICATIONS AND USAGE
LORBRENA® is indicated for the treatment of patients with anaplastic lymphoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC) whose disease has progressed on
crizotinib and at least one other ALK inhibitor for metastatic disease; or
alectinib as the first ALK inhibitor therapy for metastatic disease; or
ceritinib as the first ALK inhibitor therapy for metastatic disease.
This indication is approved under accelerated approval based on tumor response rate and duration of response [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of LORBRENA is 100 mg orally once daily, with or without food, until disease progression or unacceptable toxicity [see Clinical Pharmacology (12.3)].
Swallow tablets whole. Do not chew, crush or split tablets. Do not ingest if tablets are broken, cracked, or otherwise not intact.
Take LORBRENA at the same time each day. If a dose is missed, then take the missed dose unless the next dose is due within 4 hours. Do not take 2 doses at the same time to make up for a missed dose.
Do not take an additional dose if vomiting occurs after LORBRENA but continue with the next scheduled dose.
2.2 Dosage Modifications for Adverse Reactions
The recommended dose reductions are:
First dose reduction: LORBRENA 75 mg orally once daily
Second dose reduction: LORBRENA 50 mg orally once daily
Permanently discontinue LORBRENA in patients who are unable to tolerate 50 mg orally once daily.
Dosage modifications for adverse reactions of LORBRENA are provided in Table 1.
Table 1 Recommended LORBRENA Dosage Modifications for Adverse Reactions
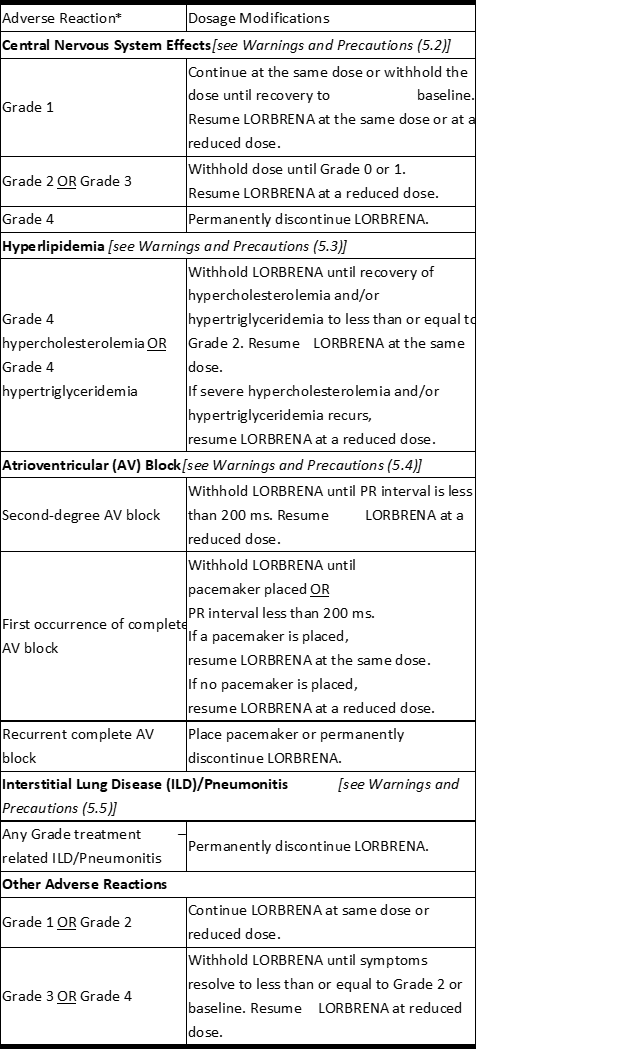
Abbreviation: AV=atrioventricular.
*Grade based on National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.
2.3 Concomitant Use of Strong or Moderate CYP3A Inducers
LORBRENA is contraindicated in patients taking strong CYP3A inducers. Discontinue strong CYP3A inducers for 3 plasma half-lives of the strong CYP3A inducer prior to initiating LORBRENA. Avoid concomitant use of LORBRENA with moderate CYP3A inducers [see Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].
2.4 Dosage Modification for Strong CYP3A Inhibitors
Avoid concomitant use of LORBRENA with strong CYP3A inhibitors. If concomitant use with a strong CYP3A inhibitor cannot be avoided, reduce the starting dose of LORBRENA from 100 mg orally once daily to 75 mg orally once daily.
In patients who have had a dose reduction to 75 mg orally once daily due to adverse reactions and who initiate a strong CYP3A inhibitor, reduce the LORBRENA dose to 50 mg orally once daily.
If concomitant use of a strong CYP3A inhibitor is discontinued, increase the LORBRENA dose (after 3 plasma half-lives of the strong CYP3A inhibitor) to the dose that was used before starting the strong inhibitor [see Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
Tablets:
25 mg: 8 mm round, tan, immediate release, film-coated, debossed with "Pfizer" on one side and "25" and "LLN" on the other side
100 mg: 8.5 mm × 17 mm oval, lavender, immediate release, film-coated, debossed with "Pfizer" on one side and "LLN 100" on the other side
4 CONTRAINDICATIONS
LORBRENA is contraindicated in patients taking strong CYP3A inducers, due to the potential for serious hepatotoxicity [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Serious Hepatotoxicity with Concomitant Use of Strong CYP3A Inducers
Severe hepatotoxicity occurred in 10 of 12 healthy subjects receiving a single dose of LORBRENA with multiple daily doses of rifampin, a strong CYP3A inducer. Grade 4 alanine aminotransferase (ALT) or aspartate aminotransferase (AST) elevations occurred in 50% of subjects, Grade 3 ALT or AST elevations occurred in 33% and Grade 2 ALT or AST elevations occurred in 8%. ALT or AST elevations occurred within 3 days and returned to within normal limits after a median of 15 days (7 to 34 days); the median time to recovery was 18 days in subjects with Grade 3 or 4 ALT or AST elevations and 7 days in subjects with Grade 2 ALT or AST elevations.
LORBRENA is contraindicated in patients taking strong CYP3A inducers. Discontinue strong CYP3A inducers for 3 plasma half-lives of the strong CYP3A inducer prior to initiating LORBRENA.
Avoid concomitant use of LORBRENA with moderate CYP3A inducers. If concomitant use of moderate CYP3A inducers cannot be avoided, monitor AST, ALT, and bilirubin 48 hours after initiating LORBRENA and at least 3 times during the first week after initiating LORBRENA.
Depending upon the relative importance of each drug, discontinue LORBRENA or the CYP3A inducer for persistent Grade 2 or higher hepatotoxicity [see Clinical Pharmacology (12.3)].
5.2 Central Nervous System Effects
A broad spectrum of central nervous system (CNS) effects can occur in patients receiving LORBRENA. These include seizures, hallucinations, and changes in cognitive function, mood (including suicidal ideation), speech, mental status, and sleep. Overall, CNS effects occurred in 54% of patients receiving LORBRENA [see Adverse Reactions (6.1)]. Cognitive effects occurred in 29% of the 332 patients who received LORBRENA at any dose in Study B7461001; 2.1% of these events were severe (Grade 3 or 4). Mood effects occurred in 24% of patients; 1.8% of these events were severe. Speech effects occurred in 14% of patients; 0.3% of these events were severe. Hallucinations occurred in 7% of patients; 0.6% of these events were severe. Mental status changes occurred in 2.1% of patients; 1.8% of these events were severe. Seizures occurred in 3% of patients, sometimes in conjunction with other neurologic findings. Sleep effects occurred in 10% of patients. The median time to first onset of any CNS effect was 1.2 months (1 day to 1.7 years). Overall, 1.5% of patients required permanent discontinuation of LORBRENA for a CNS effect; 9% required temporary discontinuation and 8% required dose reduction.
Withhold and resume at the same dose or at a reduced dose or permanently discontinue LORBRENA based on severity [see Dosage and Administration (2.2)].
5.3 Hyperlipidemia
Increases in serum cholesterol and triglycerides can occur in patients receiving LORBRENA [see Adverse Reactions (6.1)]. Grade 3 or 4 elevations in total cholesterol occurred in 17% and Grade 3 or 4 elevations in triglycerides occurred in 17% of the 332 patients who received LORBRENA in Study B7461001. The median time to onset was 15 days for both hypercholesterolemia and hypertriglyceridemia. Approximately 7% of patients required temporary discontinuation and 3% of patients required dose reduction of LORBRENA for elevations in cholesterol and in triglycerides. Eighty percent of patients required initiation of lipid-lowering medications, with a median time to onset of start of such medications of 21 days.
Initiate or increase the dose of lipid-lowering agents in patients with hyperlipidemia. Monitor serum cholesterol and triglycerides before initiating LORBRENA, 1 and 2 months after initiating LORBRENA, and periodically thereafter. Withhold and resume at the same dose for the first occurrence; resume at the same or a reduced dose of LORBRENA for recurrence based on severity [see Dosage and Administration (2.2)].
5.4 Atrioventricular Block
PR interval prolongation and atrioventricular (AV) block can occur in patients receiving LORBRENA [see Adverse Reactions (6.1), Clinical Pharmacology (12.2)]. In 295 patients who received LORBRENA at a dose of 100 mg orally once daily in Study B7461001 and who had a baseline electrocardiography (ECG), 1% experienced AV block and 0.3% experienced Grade 3 AV block and underwent pacemaker placement.
Monitor ECG prior to initiating LORBRENA and periodically thereafter. Withhold and resume at a reduced dose or at the same dose in patients who undergo pacemaker placement. Permanently discontinue for recurrence in patients without a pacemaker [see Dosage and Administration (2.2)].
5.5 Interstitial Lung Disease/Pneumonitis
Severe or life-threatening pulmonary adverse reactions consistent with interstitial lung disease (ILD)/pneumonitis can occur with LORBRENA. ILD/pneumonitis occurred in 1.5% of patients who received LORBRENA at any dose in Study B7461001, including Grade 3 or 4 ILD/pneumonitis in 1.2% of patients. One patient (0.3%) discontinued LORBRENA for ILD/pneumonitis.
Promptly investigate for ILD/pneumonitis in any patient who presents with worsening of respiratory symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, and fever). Immediately withhold LORBRENA in patients with suspected ILD/pneumonitis. Permanently discontinue LORBRENA for treatment-related ILD/pneumonitis of any severity [see Dosage and Administration (2.2)].
5.6 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, LORBRENA can cause fetal harm when administered to a pregnant woman. Administration of lorlatinib to pregnant rats and rabbits by oral gavage during the period of organogenesis resulted in malformations, increased post-implantation loss, and abortion at maternal exposures that were equal to or less than the human exposure at the recommended dose of 100 mg once daily based on area under the curve (AUC).
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use an effective non-hormonal method of contraception, since LORBRENA can render hormonal contraceptives ineffective, during treatment with LORBRENA and for at least 6 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with LORBRENA and for 3 months after the final dose [see Drug Interactions (7.2), Use in Specific Populations (8.1, 8.3), Nonclinical Toxicology (13.1)].
6 ADVERSE REACTIONS
The following adverse reactions are described elsewhere in the labeling:
Risk of Serious Hepatotoxicity with Concomitant Use of Strong CYP3A Inducers [see Warnings and Precautions (5.1)]
Central Nervous System Effects [see Warnings and Precautions (5.2)]
Hyperlipidemia [see Warnings and Precautions (5.3)]
Atrioventricular Block [see Warnings and Precautions (5.4)]
Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in Warnings and Precautions reflect exposure to LORBRENA in 332 patients with ALK-positive or ROS1-positive, metastatic non-small cell lung cancer (NSCLC) enrolled in a multi-cohort, multinational, non-comparative, dose-finding, and activity-estimating trial (Study B7461001) who received LORBRENA at doses ranging from 10 mg to 200 mg daily in single or divided doses.
The data described below reflect exposure to LORBRENA in 295 patients with ALK-positive or ROS1-positive metastatic NSCLC who received LORBRENA 100 mg orally once daily in Study B7461001. The median duration of exposure to LORBRENA was 12.5 months (1 day to 35 months) and 52% received LORBRENA for ≥12 months. Patient characteristics were a median age of 53 years (19 to 85 years), age ≥65 years (18%), female (58%), White (49%), Asian (37%), and Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1 (96%).
The most common (≥20%) adverse reactions were edema, peripheral neuropathy, cognitive effects, dyspnea, fatigue, weight gain, arthralgia, mood effects, and diarrhea; the most common (≥20%) laboratory abnormalities were hypercholesterolemia, hypertriglyceridemia, anemia, hyperglycemia, increased AST, hypoalbuminemia, increased ALT, increased lipase, and increased alkaline phosphatase.
Serious adverse reactions occurred in 32% of the 295 patients; the most frequently reported serious adverse reactions were pneumonia (3.4%), dyspnea (2.7%), pyrexia (2%), mental status changes (1.4%), and respiratory failure (1.4%). Fatal adverse reactions occurred in 2.7% of patients and included pneumonia (0.7%), myocardial infarction (0.7%), acute pulmonary edema (0.3%), embolism (0.3%), peripheral artery occlusion (0.3%), and respiratory distress (0.3%). Permanent discontinuation of LORBRENA for adverse reactions occurred in 8% of patients.
The most frequent adverse reactions that led to permanent discontinuation were respiratory failure (1.4%), dyspnea (0.7%), myocardial infarction (0.7%), cognitive effects (0.7%) and mood effects (0.7%). Approximately 48% of patients required dose interruption. The most frequent adverse reactions that led to dose interruptions were edema (7%), hypertriglyceridemia (6%), peripheral neuropathy (5%), cognitive effects (4.4%), increased lipase (3.7%), hypercholesterolemia (3.4%), mood effects (3.1%), dyspnea (2.7%), pneumonia (2.7%), and hypertension (2.0%). Approximately 24% of patients required at least 1 dose reduction for adverse reactions. The most frequent adverse reactions that led to dose reductions were edema (6%), peripheral neuropathy (4.7%), cognitive effects (4.1%), and mood effects (3.1%).
Tables 2 and 3 summarize common adverse reactions and laboratory abnormalities, respectively, in patients treated with LORBRENA in Study B7461001.
Table 2 Adverse Reactions Occurring in ≥10% of Patients in Study B7461001*

Abbreviations: NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events; SOC=System organ class.
*Adverse reactions were graded using NCI CTCAE version 4.0.
†Mood effects (including affective disorder, affect lability, aggression, agitation, anxiety, depressed mood, depression, euphoric mood, irritability, mania, mood altered, mood swings, personality change, stress, suicidal ideation).
‡Peripheral neuropathy (including burning sensation, carpal tunnel syndrome, dysesthesia, formication, gait disturbance, hypoesthesia, muscular weakness, neuralgia, neuropathy peripheral, neurotoxicity, paresthesia, peripheral sensory neuropathy, sensory disturbance).
§Cognitive effects (including events from SOC Nervous system disorders: amnesia, cognitive disorder, dementia, disturbance in attention, memory impairment, mental impairment; and also including events from SOC Psychiatric disorders: attention deficit/hyperactivity disorder, confusional state, delirium, disorientation, reading disorder).
¶Speech effects (including aphasia, dysarthria, slow speech, speech disorder)
#Sleep effects (including abnormal dreams, insomnia, nightmare, sleep disorder, sleep talking, somnambulism)
ÞVision disorder (including blindness, diplopia, photophobia, photopsia, vision blurred, visual acuity reduced, visual impairment, vitreous floaters).
ßMyalgia (including musculoskeletal pain, myalgia).
àEdema (including edema, edema peripheral, eyelid edema, face edema, generalized edema, localized edema, periorbital edema, peripheral swelling, swelling).
èFatigue (including asthenia, fatigue).
ðUpper respiratory infection (including fungal upper respiratory infection, upper respiratory infection, viral upper respiratory infection).
øRash (including dermatitis acneiform, maculopapular rash, pruritic rash, rash).
Additional clinically significant adverse reactions occurring at an incidence between 1% and 10% were hallucinations (7%).
Table 3 Worsening Laboratory Values Occurring in ≥20% of Patients in Study B7461001*
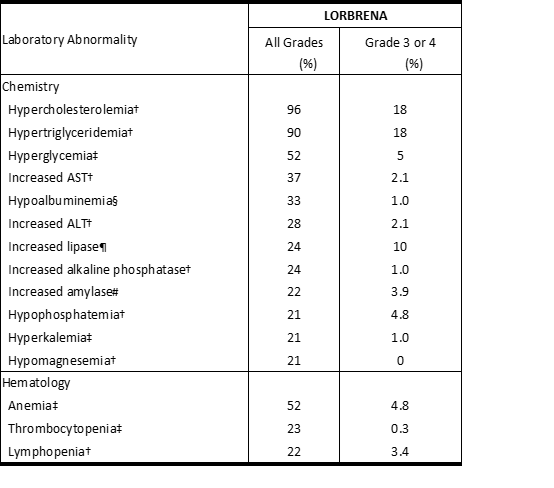
Abbreviations: ALT=alanine aminotransferase; AST=aspartate aminotransferase; NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events.
N=number of patients who had at least one on-study assessment for the parameter of interest.*Grades using NCI CTCAE version 4.0.
†N=292.
‡N=293.
§N=291.
¶N=290.
#N=284.
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on LORBRENA
Effect of CYP3A Inducers
Concomitant use of LORBRENA with a strong CYP3A inducer decreased lorlatinib plasma concentrations, which may decrease the efficacy of LORBRENA. The effect of concomitant use of LORBRENA with a moderate CYP3A inducer on lorlatinib plasma concentrations has not been studied.
Severe hepatotoxicity occurred in healthy subjects receiving LORBRENA with rifampin, a strong CYP3A inducer. In 12 healthy subjects receiving a single 100 mg dose of LORBRENA with multiple daily doses of rifampin, Grade 3 or 4 increases in ALT or AST occurred in 83% of subjects and Grade 2 increases in ALT or AST occurred in 8%. A possible mechanism for hepatotoxicity is through activation of the pregnane X receptor (PXR) by LORBRENA and rifampin, which are both PXR agonists. The risk of hepatotoxicity with concomitant use of LORBRENA and moderate CYP3A inducers that are also PXR agonists is unknown.
LORBRENA is contraindicated in patients taking strong CYP3A inducers. Discontinue strong CYP3A inducers for 3 plasma half-lives of the strong CYP3A inducer prior to initiating LORBRENA.
Avoid concomitant use of LORBRENA with moderate CYP3A inducers. If concomitant use of moderate CYP3A inducers cannot be avoided, monitor ALT, AST, and bilirubin as recommended [see Dosage and Administration (2.3), Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].
Effect of Strong CYP3A Inhibitors
Concomitant use with a strong CYP3A inhibitor increased lorlatinib plasma concentrations, which may increase the incidence and severity of adverse reactions of LORBRENA. Avoid the concomitant use of LORBRENA with a strong CYP3A inhibitor. If concomitant use cannot be avoided, reduce LORBRENA dose as recommended [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
7.2 Effect of LORBRENA on Other Drugs
CYP3A Substrates
Concomitant use of LORBRENA decreases the concentration of CYP3A substrates [see Clinical Pharmacology (12.3)], which may reduce the efficacy of these substrates. Avoid concomitant use of LORBRENA with CYP3A substrates, where minimal concentration changes may lead to serious therapeutic failures. If concomitant use is unavoidable, increase the CYP3A substrate dosage in accordance with approved product labeling.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], LORBRENA can cause embryo-fetal harm when administered to a pregnant woman. There are no available data on LORBRENA use in pregnant women. Administration of lorlatinib to pregnant rats and rabbits by oral gavage during the period of organogenesis resulted in malformations, increased post-implantation loss, and abortion at maternal exposures that were equal to or less than the human exposure at the recommended dose of 100 mg once daily based on AUC (see Data). Advise a pregnant woman of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Preliminary embryo-fetal development studies investigating the administration of lorlatinib during the period of organogenesis were conducted in rats and rabbits. In rabbits, lorlatinib administration resulted in abortion and total loss of pregnancy at doses of 15 mg/kg (approximately 3 times the human exposure at the recommended dose of 100 mg) or greater. At a dose of 4 mg/kg (approximately 0.6 times the human exposure at the recommended dose of 100 mg) toxicities included increased post-implantation loss and malformations including rotated limbs, malformed kidneys, domed head, high arched palate, and dilation of the cerebral ventricles. In rats, administration of lorlatinib resulted in total loss of pregnancy at doses of 4 mg/kg (approximately 5 times the human exposure at the recommended dose of 100 mg) or greater. At a dose of 1 mg/kg (approximately equal to the human exposure at the recommended dose of 100 mg) there was increased post-implantation loss, decreased fetal body weight, and malformations including gastroschisis, rotated limbs, supernumerary digits, and vessel abnormalities.
8.2 Lactation
Risk Summary
There are no data on the presence of lorlatinib or its metabolites in either human or animal milk or its effects on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in breastfed infants, instruct women not to breastfeed during treatment with LORBRENA and for 7 days after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating LORBRENA [see Use in Specific Populations (8.1)].
Contraception
LORBRENA can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise female patients of reproductive potential to use effective non-hormonal contraception during treatment with LORBRENA and for at least 6 months after the final dose. Advise females of reproductive potential to use a non-hormonal method of contraception, because LORBRENA can render hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with LORBRENA and for at least 3 months after the final dose [see Nonclinical Toxicology (13.1)].
Infertility
Males
Based on findings from animal studies, LORBRENA may transiently impair male fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of LORBRENA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 295 patients in Study B7461001 who received 100 mg LORBRENA orally once daily, 18% of patients were aged 65 years or older. Although data are limited, no clinically important differences in safety or efficacy were observed between patients aged 65 years or older and younger patients.
8.6 Hepatic Impairment
No dose adjustment is recommended for patients with mild hepatic impairment (total bilirubin ≤ upper limit of normal [ULN] with AST > ULN or total bilirubin >1 to 1.5 × ULN with any AST). The recommended dose of LORBRENA has not been established for patients with moderate or severe hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dose adjustment is recommended for patients with mild or moderate renal impairment (creatinine clearance [CLcr] 30 to 89 mL/min estimated by Cockcroft-Gault). The recommended dose of LORBRENA has not been established for patients with severe renal impairment [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
LORBRENA (lorlatinib) is a kinase inhibitor for oral administration. The molecular formula is C21H19FN6O2 (anhydrous form) and the molecular weight is 406.41 Daltons. The chemical name is (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-4,8-methenopyrazolo[4,3-h][2,5,11] benzoxadiazacyclotetradecine-3-carbonitrile. The chemical structure is shown below:
Lorlatinib is a white to off-white powder with a pKa of 4.92. The solubility of lorlatinib in aqueous media decreases over the range pH 2.55 to pH 8.02 from 32.38 mg/mL to 0.17 mg/mL. The log of the distribution coefficient (octanol/water) at pH 9 is 2.45.
LORBRENA is supplied as tablets containing 25 mg or 100 mg of lorlatinib with the following inactive ingredients: microcrystalline cellulose, dibasic calcium phosphate anhydrous, sodium starch glycolate, and magnesium stearate. The film-coating contains hydroxypropyl methylcellulose (HPMC) 2910/hypromellose, lactose monohydrate, macrogol/polyethylene glycol (PEG) 3350, triacetin, titanium dioxide, ferrosoferric oxide/black iron oxide, and iron oxide red.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lorlatinib is a kinase inhibitor with in vitro activity against ALK and ROS1 as well as TYK1, FER, FPS, TRKA, TRKB, TRKC, FAK, FAK2, and ACK. Lorlatinib demonstrated in vitro activity against multiple mutant forms of the ALK enzyme, including some mutations detected in tumors at the time of disease progression on crizotinib and other ALK inhibitors.
In mice subcutaneously implanted with tumors harboring EML4 fusions with either ALK variant 1 or ALK mutations, including the G1202R and I1171T mutations detected in tumors at the time of disease progression on ALK inhibitors, administration of lorlatinib resulted in antitumor activity. Lorlatinib also demonstrated anti-tumor activity and prolonged survival in mice implanted intracranially with EML4-ALK-driven tumor cell lines. The overall antitumor activity of lorlatinib in in vivo models was dose-dependent and correlated with inhibition of ALK phosphorylation.
12.2 Pharmacodynamics
Exposure-Response Relationships
Based on the data from Study B7461001, exposure-response relationships for Grade 3 or 4 hypercholesterolemia and for any Grade 3 or 4 adverse reaction were observed at steady-state exposures achieved at the recommended dosage, with higher probability of the occurrence of adverse reactions with increasing lorlatinib exposure.
Cardiac Electrophysiology
In 295 patients who received LORBRENA at the recommended dosage of 100 mg once daily and had an ECG measurement in Study B7461001, the maximum mean change from baseline for PR interval was 16.4 ms (2-sided 90% upper confidence interval [CI] 19.4 ms). Among the 284 patients with PR interval <200 ms at baseline, 14% had PR interval prolongation ≥200 ms after starting LORBRENA. The prolongation of PR interval occurred in a concentration-dependent manner. Atrioventricular block occurred in 1% of patients.
In 275 patients who received LORBRENA at the recommended dosage in the activity-estimating portion of Study B7461001, no large mean increases from baseline in the QTcF interval (i.e., >20 ms) were detected.
12.3 Pharmacokinetics
Steady-state lorlatinib maximum plasma concentration (Cmax) increases proportionally and AUC increased slightly less than proportionally over the dose range of 10 mg to 200 mg orally once daily (0.1 to 2 times the recommended dosage). At the recommended dosage, the mean (coefficient of variation [CV] %) Cmax was 577 ng/mL (42%) and the AUC0–24h was 5650 ng∙h/mL (39%) in patients with cancer. Lorlatinib oral clearance increased at steady-state compared to single dose, indicating autoinduction.
Absorption
The median lorlatinib Tmax was 1.2 hours (0.5 to 4 hours) following a single oral 100 mg dose and 2 hours (0.5 to 23 hours) following 100 mg orally once daily at steady state.
The mean absolute bioavailability is 81% (90% CI 75.7%, 86.2%) after oral administration compared to intravenous administration.
Effect of Food
Administration of LORBRENA with a high fat, high calorie meal (approximately 1000 calories with 150 calories from protein, 250 calories from carbohydrate, and 500 to 600 calories from fat) had no clinically meaningful effect on lorlatinib pharmacokinetics.
Distribution
In vitro, lorlatinib was 66% bound to plasma proteins at a concentration of 2.4 µM. The blood-to-plasma ratio was 0.99. The mean (CV%) steady state volume of distribution (Vss) was 305 L (28%) following a single intravenous dose.
Elimination
The mean plasma half-life (t½) of lorlatinib was 24 hours (40%) after a single oral 100 mg dose of LORBRENA. The mean oral clearance (CL/F) was 11 L/h (35%) following a single oral 100 mg dose and increased to 18 L/h (39%) at steady state, suggesting autoinduction.
Metabolism
In vitro, lorlatinib is metabolized primarily by CYP3A4 and UGT1A4, with minor contribution from CYP2C8, CYP2C19, CYP3A5, and UGT1A3.
In plasma, a benzoic acid metabolite (M8) of lorlatinib resulting from the oxidative cleavage of the amide and aromatic ether bonds of lorlatinib accounted for 21% of the circulating radioactivity in a human [14C] mass balance study. The oxidative cleavage metabolite, M8, is pharmacologically inactive.
Excretion
Following a single oral 100 mg dose of radiolabeled lorlatinib, 48% of the radioactivity was recovered in urine (<1% as unchanged) and 41% in feces (about 9% as unchanged).
Specific Populations
No clinically meaningful differences in lorlatinib pharmacokinetics were observed based on age (19 to 85 years), sex, race/ethnicity, body weight, mild to moderate renal impairment (CLcr 30 to 89 mL/min), mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin > 1.5 × ULN and any AST), or metabolizer phenotypes for CYP3A5 and CYP2C19. The effect of moderate to severe hepatic impairment or severe renal impairment on lorlatinib pharmacokinetics is unknown [see Use in Specific Populations (8.6, 8.7)].
Drug Interaction Studies
Clinical Studies
Effect of CYP3A Inducers on Lorlatinib: Twelve healthy subjects received rifampin, a strong CYP3A inducer that also activates PXR, 600 mg once daily for 8 days (Days 1 to 8) and a single oral 100 mg dose of LORBRENA on Day 8. The coadministration of rifampin with LORBRENA reduced the mean lorlatinib AUCinf by 85% and Cmax by 76%. Grade 2 to 4 increases in ALT or AST occurred within 3 days. Grade 4 ALT or AST elevations occurred in 50%, Grade 3 ALT or AST elevations in 33%, and Grade 2 ALT or AST elevations occurred in 8% of subjects. ALT and AST returned to within normal limits within 7 to 34 days (median 15 days). The effect of the concomitant use of moderate CYP3A inducers on lorlatinib pharmacokinetics or the risk of hepatotoxicity with the concomitant use of moderate CYP3A inducers is unknown [see Drug Interactions (7.1)].
Effect of Strong CYP3A Inhibitors on Lorlatinib: Itraconazole, a strong CYP3A inhibitor, increased AUCinf by 42% and increased Cmax by 24% of a single oral 100 mg dose of LORBRENA [see Drug Interactions (7.1)].
Effect of Lorlatinib on CYP3A Substrates: LORBRENA 150 mg orally once daily for 15 days decreased AUCinf by 64% and Cmax by 50% of a single oral 2 mg dose of midazolam (a sensitive CYP3A substrate) [see Drug Interactions (7.2)].
Effect of Acid-Reducing Agents on Lorlatinib: Concomitant use of a proton pump inhibitor, rabeprazole, did not have a clinically meaningful effect on lorlatinib pharmacokinetics.
In Vitro Studies
Effect of Lorlatinib on CYP Enzymes: In vitro studies indicate that lorlatinib is a time-dependent inhibitor as well as an inducer of CYP3A and that it activates PXR, with the net effect in vivo being induction. Lorlatinib induces CYP2B6 and activates the human constitutive androstane receptor (CAR). Lorlatinib and the major circulating metabolite, M8, do not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6. M8 does not inhibit CYP3A.
M8 does not induce CYP1A2, CYP2B6, and CYP3A.
Effects of Lorlatinib on UDP-glucuronosyltransferase (UGT): Lorlatinib and M8 do not inhibit UGT1A1, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15.
Effect of Lorlatinib on Transporters: Lorlatinib inhibits P-glycoprotein (P-gp), organic cation transporter (OCT)1, organic anion transporter (OAT)3, multidrug and toxin extrusion (MATE)1, and intestinal breast cancer resistance protein (BCRP). Lorlatinib does not inhibit organic anion transporting polypeptide (OATP)1B1, OATP1B3, OAT1, OCT2, MATE2K, and systemic BCRP. M8 does not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1, and MATE2K.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with lorlatinib. Lorlatinib was aneugenic in an in vitro assay in human lymphoblastoid TK6 cells and positive for micronuclei formation in vivo in the bone marrow of rats. Lorlatinib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay.
Dedicated fertility studies were not conducted with lorlatinib. Findings in male reproductive organs occurred in repeat-dose toxicity studies and included lower testicular, epididymal, and prostate weights; testicular tubular degeneration/atrophy; prostatic atrophy; and/or epididymal inflammation at 15 mg/kg/day and 7 mg/kg/day in rats and dogs, respectively (approximately 8 and 2 times, respectively, the human exposure at the recommended dose of 100 mg based on AUC). The effects on male reproductive organs were reversible.
13.2 Animal Toxicology and/or Pharmacology
Distended abdomen, skin rash, and increased cholesterol and triglycerides occurred in animals. These findings were accompanied by hyperplasia and dilation of the bile ducts in the liver and acinar atrophy of the pancreas in rats at 15 mg/kg/day and in dogs at 2 mg/kg/day (approximately 8 and 0.5 times, respectively, the human exposure at the recommended dose of 100 mg based on AUC). All effects were reversible within the recovery period.
14 CLINICAL STUDIES
14.1 ALK-Positive Metastatic NSCLC Previously Treated with an ALK Kinase Inhibitor
The efficacy of LORBRENA was demonstrated in a subgroup of patients with ALK-positive metastatic non-small cell lung cancer (NSCLC) previously treated with one or more ALK kinase inhibitors who were enrolled in a non-randomized, dose-ranging and activity-estimating, multi-cohort, multicenter study (Study B7461001; NCT01970865). Patients included in this subgroup were required to have metastatic disease with at least 1 measurable target lesion according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (v1.1), ECOG performance status of 0 to 2, and documented ALK rearrangement in tumor tissue as determined by fluorescence in situ hybridization (FISH) assay or by Immunohistochemistry (IHC), and received LORBRENA 100 mg orally once daily. Patients with asymptomatic CNS metastases, including patients with stable or decreasing steroid use within 2 weeks prior to study entry, were eligible. Patients with severe, acute, or chronic psychiatric conditions including suicidal ideation or behavior were excluded. In addition, for patients with ALK-positive metastatic NSCLC, the extent and type of prior treatment was specified for each individual cohort (see Table 4). The major efficacy outcome measures were overall response rate (ORR) and intracranial ORR, according to RECIST v1.1, as assessed by Independent Central Review (ICR) committee. Data were pooled across all subgroups listed in Table 4. Additional efficacy outcome measures included duration of response (DOR), and intracranial DOR.
A total of 215 patients were enrolled across the subgroups in Table 4. The distribution of patients by type and extent of prior therapy is provided in Table 4. The demographic characteristics across all 215 patients were: 59% female, 51% White, 34% Asian, and the median age was 53 years (29 to 85 years) with 18% of patients ≥65 years. The ECOG performance status at baseline was 0 or 1 in 96% of patients. All patients had metastatic disease and 95% had adenocarcinoma. Brain metastases as identified by ICR were present in 69% of patients; of these, 60% had received prior radiation to the brain and 60% (n=89) had measurable disease per ICR.
Table 4 Extent of Prior Therapy in the Subgroup of Patients with Previously Treated ALK-Positive Metastatic NSCLC in Study B7461001
Abbreviations: ALK=anaplastic lymphoma kinase; NSCLC=non-small cell lung cancer.
*Chemotherapy administered in the metastatic setting.
Efficacy results for Study B7461001 are summarized in Tables 5 and 6.
Table 5 Efficacy Results in Study B7461001 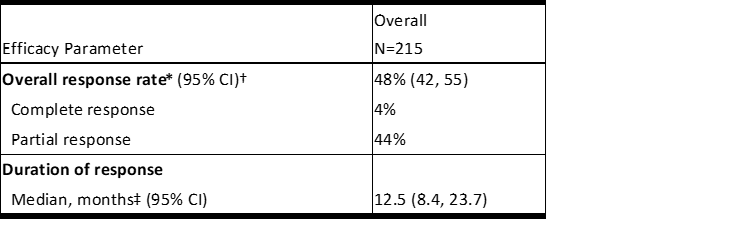
Abbreviations: CI=confidence interval; N=number of patients.
*Per Independent Central Review.
†Using exact method based on binomial distribution.
‡Estimated using the Kaplan Meier method.
An assessment of intracranial ORR and the duration of response for CNS metastases in the subgroup of 89 patients in Study B7461001 with baseline measurable lesions in the CNS according to RECIST v1.1 are summarized in Table 6. Of these, 56 (63%) patients received prior brain radiation, including 42 patients (47%) who completed brain radiation treatment at least 6 months before starting treatment with LORBRENA.
Table 6 Intracranial Response Rate in Patients with Measurable Intracranial Lesions in Study B7461001 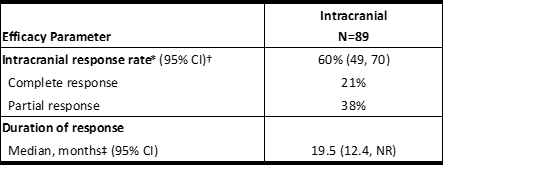
Abbreviations: CI=confidence interval; N=number of patients; NR=not reached.
*Per Independent Central Review.
†Using exact method based on binomial distribution.
‡Estimated using the Kaplan-Meier method.
In exploratory analyses conducted in subgroups defined by prior therapy, the response rates to LORBRENA were:
- ORR = 39% (95% CI: 30, 48) in 119 patients who received crizotinib and at least one other ALK inhibitor, with or without prior chemotherapy
- ORR = 31% (95% CI: 9, 61) in 13 patients who received alectinib as their only ALK inhibitor, with or without prior chemotherapy
- ORR = 46% (95% CI: 19, 75) in 13 patients who received ceritinib as their only ALK inhibitor, with or without prior chemotherapy
16 HOW SUPPLIED/STORAGE AND HANDLING
Table 7 describes the available strengths and package configurations for LORBRENA:
Table 7 LORBRENA Tablets 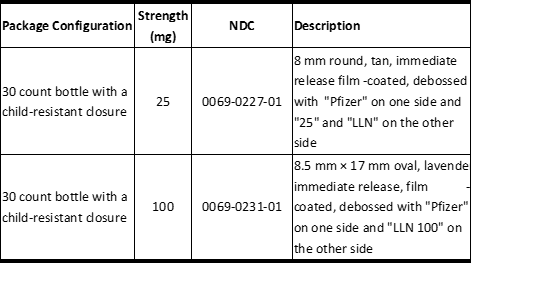
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Risk of Serious Hepatotoxicity with Concomitant Use of Strong CYP3A Inducers
Inform patients of the potential risk of hepatoxicity with the concomitant use of strong CYP3A inducers.
Advise patients to inform their healthcare providers of all medications they are taking, including prescription medicines, over-the-counter drugs, vitamins, and herbal products (e.g., St. John's wort) [see Warnings and Precautions (5.1)].
Central Nervous System (CNS) Effects
Advise patients to notify their healthcare provider if they experience new or worsening CNS symptoms [see Warnings and Precautions (5.2)].
Hyperlipidemia
Inform patients that serum cholesterol and triglycerides will be monitored during treatment. Advise patients that initiation or an increase in the dose of lipid-lowering agents may be required [see Warnings and Precautions (5.3)].
Atrioventricular (AV) Block
Inform patients of the risks of AV block. Advise patients to contact their healthcare provider immediately to report new or worsening cardiac symptoms [see Warnings and Precautions (5.4)].
Interstitial Lung Disease (ILD)/Pneumonitis
Inform patients of the risks of severe ILD/pneumonitis. Advise patients to contact their healthcare provider immediately to report new or worsening respiratory symptoms [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective non-hormonal contraception during treatment with LORBRENA and for at least 6 months after the final dose [see Use in Specific Populations (8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with LORBRENA and for at least 3 months after the final dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
Advise women not to breastfeed during treatment with LORBRENA and for 7 days after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise males of reproductive potential that LORBRENA may transiently impair fertility [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
This product's label may have been updated. For full prescribing information, please visit www.LORBRENA.com.

LAB-1162-1.0
PATIENT INFORMATION
LORBRENA (lor-BREN-ah)
(lorlatinib)
tabletsWhat is the most important information I should know about LORBRENA?
LORBRENA may cause serious side effects, including:
- Liver problems due to interactions with other medicines. It is important to know what medicines should not be taken with LORBRENA.
- Problems with brain (central nervous system [CNS]) function. Many patients experienced problems with brain function including problems with thinking (such as forgetfulness or confusion), mood (such as depression), speech, seeing or hearing things that are not real (hallucinations), and seizures during treatment with LORBRENA. In some patients, these problems are severe and your healthcare provider may need to have you stop taking LORBRENA.
- Increases in the cholesterol and triglycerides (lipid) levels in your blood. Most patients will have an increase in the lipid levels in your blood during treatment with LORBRENA.
- If you have increases in the lipid levels in your blood during treatment with LORBRENA, your healthcare provider may need to start you on a medicine to lower the levels. If you are already taking a medicine to lower the lipid levels in your blood, your healthcare provider may need to increase your dose of that medicine.
- Your healthcare provider should do blood tests to check the lipid levels in your blood before starting treatment, 1 to 2 months after starting treatment, and during treatment with LORBRENA.
- Heart problems. LORBRENA may cause very slow or abnormal heartbeats. Your healthcare provider should check your heart rhythm (electrocardiogram [EKG]) before starting and during treatment with LORBRENA. Tell your healthcare provider right away if you feel dizzy or faint or have abnormal heartbeats. In some patients, these problems are severe and your healthcare provider may need to have you stop taking LORBRENA or have a pacemaker placed.
- Lung problems. LORBRENA may cause severe or life-threatening swelling (inflammation) of the lungs during treatment that can lead to death. Symptoms may be similar to those from lung cancer. Tell your healthcare provider right away if you have any new or worsening symptoms of lung problems, including trouble breathing, shortness of breath, cough, or fever.
In some patients, these problems are severe and your healthcare provider may need to have you stop taking LORBRENA. See "What are possible side effects of LORBRENA?" for more information about side effects.
What is LORBRENA?
LORBRENA is a prescription medicine that is used to treat people with non-small cell lung cancer (NSCLC)
- that is caused by an abnormal anaplastic lymphoma kinase (ALK) gene and,
- that has spread to other parts of your body and,
- who have taken the medicine alectinib or ceritinib or who have taken both the medicine crizotinib and at least 1 other medicine to treat NSCLC that is caused by the ALK gene, and
- their NSCLC is no longer responding to these treatments.
It is not known if LORBRENA is safe and effective in children.
Do not take LORBRENA if you take certain other medicines called strong CYP3A inducers. Ask your healthcare provider for a list of these medicines if you are not sure.
Before taking LORBRENA, tell your healthcare provider about all of your medical conditions, including if you:
- are taking other medications
- have had episodes of depression or seizures
- have high levels of cholesterol or triglycerides in your blood
- have problems with your heart beat
- have lung or breathing problems
- are pregnant or plan to become pregnant. LORBRENA can harm your unborn baby.
- Your healthcare provider will do a pregnancy test before you start treatment with LORBRENA.
- Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with LORBRENA.
–
Females who are able to become pregnant should use effective non-hormonal birth control during treatment with LORBRENA and for at least 6 months after the final dose of LORBRENA. Birth control pills (oral contraceptives) and other hormonal forms of birth control may not be effective if used during treatment with LORBRENA. Talk to your healthcare provider about birth control choices that are right for you during this time.
–
Males who have female partners who are able to become pregnant should use effective birth control during treatment with LORBRENA and for at least 3 months after the final dose of LORBRENA.
- are breastfeeding or plan to breastfeed. It is not known if LORBRENA passes into your breast milk. Do not breastfeed during treatment with LORBRENA and for 7 days after the final dose. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements.
How should I take LORBRENA?
- Take LORBRENA exactly as your healthcare provider tells you to take it. Do not change your dose or stop taking LORBRENA unless your healthcare provider tells you to.
- Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with LORBRENA if you develop side effects.
- Swallow LORBRENA tablets whole. Do not chew, crush, or split LORBRENA tablets. Do not take LORBRENA tablets if they are broken, cracked, or not intact.
- Take LORBRENA at approximately the same time each day.
- You may take LORBRENA with or without food.
- If you miss a dose, take it as soon as you remember. However, if it is close to the time of your next dose (within 4 hours), just take your next dose at your regular time.
- If you vomit after taking a dose of LORBRENA, do not take an extra dose. Take your next dose at your regular time.
What are the possible side effects of LORBRENA?
The most common side effects of LORBRENA include:
- swelling in your arms, legs, hands and feet (edema)
- numbness and tingling feeling in your joints or arms and legs (peripheral neuropathy)
- difficulty thinking or confusion
- difficulty breathing
- tiredness (fatigue)
- weight gain
- pain in your joints
- changes in mood, feeling sad or anxious
- diarrhea
LORBRENA may cause decreased fertility in males. In males, this could affect your ability to father a child. Talk to your healthcare provider if you have concerns about fertility.
These are not all of the possible side effects of LORBRENA. For more information, ask your healthcare provider or pharmacist.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store LORBRENA?
- Store LORBRENA at room temperature between 68°F to 77°F (20°C to 25°C).
Keep LORBRENA and all medicines out of the reach of children.
General information about the safe and effective use of LORBRENA
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use LORBRENA for a condition for which it was not prescribed. Do not give LORBRENA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about LORBRENA that is written for health professionals.
What are the ingredients in LORBRENA?
Active ingredient: lorlatinib
Inactive ingredients: microcrystalline cellulose, dibasic calcium phosphate anhydrous, sodium starch glycolate, and magnesium stearate.
Film-coating contains: hydroxypropyl methylcellulose (HPMC) 2910/hypromellose, lactose monohydrate, macrogol/ polyethylene glycol (PEG) 3350, triacetin, titanium dioxide, ferrosoferric oxide/black iron oxide, and iron oxide red.
For more information, go to www.LORBRENA.com.

LAB-1163-1.0
This Patient Information has been approved by the U.S. Food and Drug Administration. November 2018
PRINCIPAL DISPLAY PANEL - 25 mg Tablet Bottle Label
Pfizer
NDC 0069-0227-01
Lorbrena®
(lorlatinib) tablets
25 mg30 Tablets
Rx only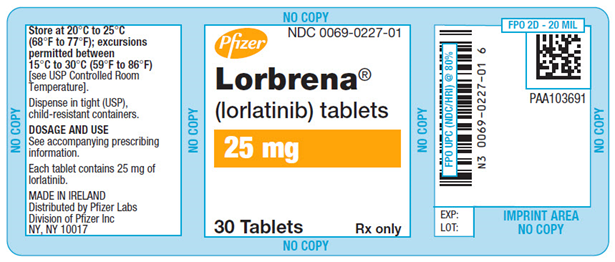
PRINCIPAL DISPLAY PANEL - 100 mg Tablet Bottle Label
Pfizer
NDC 0069-0231-01
Lorbrena®
(lorlatinib) tablets100 mg
30 Tablets
Rx only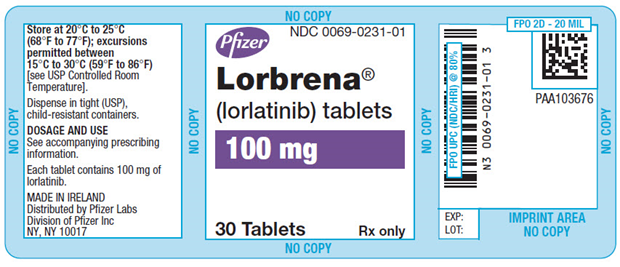
【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
-

通用名: 洛拉替尼片
商品名: Lorbrexen
规格: 100mg×30片
产地: 孟加拉珠峰制药(Everest Pharmaceuticals Ltd.)
国际参考零售价:¥**/盒
-